Constraint contains parent classes for all constraints. A Constraint is used to set certain rules for the stochastic engine to evolve the atomic system. Therefore it has become possible to fully customize and set any possibly imaginable rule.
fullrmc.Core.Constraint.ConstraintBases: fullrmc.Core.Collection.ListenerBase
A constraint is used to direct the evolution of the atomic configuration towards the desired and most meaningful one.
constraintIdConstraint unique ID create at instanciation time.
constraintNameConstraints unique name in engine given when added to engine.
engineStochastic fullrmc’s engine instance.
usedFrameGet used frame in engine. If None then engine is not defined yet
computationCostComputation cost number.
stateConstraint’s state.
triedConstraint’s number of tried moves.
acceptedConstraint’s number of accepted moves.
usedConstraint’s used flag. Defines whether constraint is used in the stochastic engine at runtime or set inactive.
varianceSquaredConstraint’s varianceSquared used in the stochastic engine at runtime to calculate the total constraint’s standard error.
standardErrorConstraint’s standard error value.
originalDataConstraint’s original data calculated upon initialization.
dataConstraint’s current calculated data.
activeAtomsDataBeforeMoveConstraint’s current calculated data before last move.
activeAtomsDataAfterMoveConstraint’s current calculated data after last move.
afterMoveStandardErrorConstraint’s current calculated StandardError after last move.
amputationDataConstraint’s current calculated data after amputation.
amputationStandardErrorConstraint’s current calculated StandardError after amputation.
multiframeWeightGet constraint weight towards total in a multiframe system.
is_in_engine(engine)Get whether constraint is already in defined and added to engine. It can be the same exact instance or a repository pulled instance of the same constraintId
| Parameters: |
|
|---|---|
| Returns: |
|
set_variance_squared(value, frame=None)Set constraint’s variance squared that is used in the computation of the total stochastic engine standard error.
| Parameters: |
|
|---|
set_computation_cost(value, frame=None)Set constraint’s computation cost value. This is used at stochastic engine runtime to minimize computations and enhance performance by computing less costly constraints first. At every step, constraints will be computed in order starting from the less to the most computationally costly. Therefore upon rejection of a step because of an unsatisfactory rigid constraint, the left un-computed constraints at this step are guaranteed to be the most time coslty ones.
| Parameters: |
|
|---|
set_used(*args, **kwargs)Set used flag.
| Parameters: |
|
|---|
set_state(value)Set constraint’s state. When constraint’s state and stochastic engine’s state don’t match, constraint’s data must be re-calculated.
| Parameters: |
|
|---|
set_tried(value)Set constraint’s number of tried moves.
| Parameters: |
|
|---|
increment_tried()Increment number of tried moves.
set_accepted(value)Set constraint’s number of accepted moves.
| Parameters: |
|
|---|
increment_accepted()Increment constraint’s number of accepted moves.
set_standard_error(value)Set constraint’s standardError value.
| Parameters: |
|
|---|
set_data(value)Set constraint’s data value.
| Parameters: |
|
|---|
set_active_atoms_data_before_move(value)Set constraint’s before move happens active atoms data value.
| Parameters: |
|
|---|
set_active_atoms_data_after_move(value)Set constraint’s after move happens active atoms data value.
| Parameters: |
|
|---|
set_after_move_standard_error(value)Set constraint’s standard error value after move happens.
| Parameters: |
|
|---|
set_amputation_data(value)Set constraint’s after amputation data.
| Parameters: |
|
|---|
set_amputation_standard_error(value)Set constraint’s standardError after amputation.
| Parameters: |
|
|---|
reset_constraint(reinitialize=True, flags=False, data=False, frame=None)Reset constraint.
| Parameters: |
|
|---|
update_standard_error()Compute and set constraint’s standard error by calling compute_standard_error method and passing constraint’s data.
get_constraints_properties(frame)Get a dictionary look up table of constraint’s properties
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraint_value()Method must be overloaded in children classes.
get_constraint_original_value()Method must be overloaded in children classes.
compute_standard_error()Method must be overloaded in children classes.
compute_data(update=True)Method must be overloaded in children classes.
compute_before_move(realIndexes, relativeIndexes)Method must be overloaded in children classes.
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)Method must be overloaded in children classes.
accept_move(realIndexes, relativeIndexes)Method must be overloaded in children classes.
reject_move(realIndexes, relativeIndexes)Method must be overloaded in children classes.
compute_as_if_amputated(realIndex, relativeIndex)Method must be overloaded in children classes.
compute_as_if_inserted(realIndex, relativeIndex)Method must be overloaded in children classes.
accept_amputation(realIndex, relativeIndex)Method must be overloaded in children classes.
reject_amputation(realIndex, relativeIndex)Method must be overloaded in children classes.
accept_insertion(realIndex, relativeIndex)Method must be overloaded in children classes.
reject_insertion(realIndex, relativeIndex)Method must be overloaded in children classes.
export(fileName, frame=None, format='%s', delimiter='\t', comments='#')Export constraint data to text file or to an archive of files.
| Parameters: |
|
|---|
plot(frame=None, axes=None, subAdParams={'bottom': None, 'hspace': 0.4, 'left': None, 'right': None, 'top': None, 'wspace': None}, dataParams={'label': 'Y', 'linewidth': 2}, xlabelParams={'size': 10, 'xlabel': 'Core-Shell atoms'}, ylabelParams={'size': 10, 'ylabel': 'Coordination number'}, xticksParams={'fontsize': 8, 'rotation': 90}, yticksParams={'fontsize': 8, 'rotation': 0}, legendParams={'fontsize': 8, 'frameon': False, 'loc': 'upper right', 'ncol': 1}, titleParams={'fontsize': 10, 'label': '@{frame} (${numberOfRemovedAtoms:.1f}$ $rem.$ $at.$) $Std.Err.={standardError:.3f}$ - $used$ $({used})$'}, gridParams=None, show=True, **paramsKwargs)Plot constraint data. This can be overloaded in children classes.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Constraint.ExperimentalConstraint(experimentalData, dataWeights=None, scaleFactor=1.0, adjustScaleFactor=(0, 0.8, 1.2))Bases: fullrmc.Core.Constraint.Constraint
Experimental constraint is any constraint related to experimental data.
| Parameters: |
|
|---|
NB: If adjustScaleFactor first item (frequency) is 0, the scale factor will remain untouched and the limits minimum and maximum won’t be checked.
experimentalDataExperimental data of the constraint.
dataWeightsExperimental data points weight
multiframeWeightGet constraint weight towards total in a multiframe system.
multiframePriorGet constraint multiframe prior array.
scaleFactorConstraint’s scaleFactor.
adjustScaleFactorAdjust scale factor tuple.
adjustScaleFactorFrequencyScale factor adjustment frequency.
adjustScaleFactorMinimumScale factor adjustment minimum number allowed.
adjustScaleFactorMaximumScale factor adjustment maximum number allowed.
limitsUsed data X limits.
limitsIndexStartUsed data start index as calculated from limits.
limitsIndexEndUsed data end index as calculated from limits.
set_scale_factor(scaleFactor)Set the scale factor. This method doesn’t allow specifying frames. It will target used frame only.
| Parameters: |
|
|---|
set_adjust_scale_factor(adjustScaleFactor)Set adjust scale factor. This method doesn’t allow specifying frames. It will target used frame only.
| Parameters: |
|
|---|
set_experimental_data(experimentalData)Set the constraint’s experimental data. This method will raise an error if called after adding constraint to stochastic engine.
| Parameters: |
|
|---|
set_data_weights(dataWeights, frame=None)Set experimental data points weight. Data weights will be automatically normalized.
| Parameters: |
|
|---|
check_experimental_data(experimentalData)Checks the constraint’s experimental data This method must be overloaded in all experimental constraint sub-classes.
| Parameters: |
|
|---|
fit_scale_factor(experimentalData, modelData, dataWeights)The best scale factor value is computed by minimizing \(E=sM\).
This method doesn’t allow specifying frames. It will target used frame only.
| Parameters: |
|
|---|---|
| Returns: |
|
NB: This method won’t update the internal scale factor value of the constraint. It always computes the best scale factor given experimental and atomic model data.
get_adjusted_scale_factor(experimentalData, modelData, dataWeights)Checks if scale factor should be updated according to the given scale factor frequency and engine’s accepted steps. If adjustment is due, a new scale factor will be computed using fit_scale_factor method, otherwise the the constraint’s scale factor will be returned.
| Parameters: |
|
|---|---|
| Returns: | #. scaleFactor (number): Constraint’s scale factor or the new scale factor fit value. |
NB: This method WILL NOT UPDATE the internal scale factor value of the constraint.
compute_standard_error(experimentalData, modelData)Compute the squared deviation between modal computed data and the experimental ones.
Where:
\(N\) is the total number of experimental data points.
\(W_{i}\) is the data point weight. It becomes equivalent to 1 when dataWeights is set to None.
\(Y(X_{i})\) is the experimental data point \(X_{i}\).
\(F(X_{i})\) is the computed from the model data \(X_{i}\).
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraints_properties(frame)Get a dictionary look up table of constraint’s properties that are needed to plot or export
| Parameters: |
|
|---|---|
| Returns: |
|
plot(frame=None, axes=None, asMesoscopic=False, intra=True, inter=True, shapeFunc=True, subAdParams={'bottom': None, 'hspace': 0.4, 'left': None, 'right': None, 'top': None, 'wspace': None}, totParams={'color': 'black', 'label': 'total', 'linewidth': 3.0, 'zorder': 1}, expParams={'color': 'red', 'label': 'experimental', 'marker': 'o', 'markersize': 7.5, 'markevery': 1, 'zorder': 0}, noWParams={'color': 'black', 'label': 'total - no window', 'linewidth': 1.0, 'zorder': 1}, shaParams={'color': 'black', 'label': 'shape function', 'linestyle': 'dashed', 'linewidth': 1.0, 'zorder': 2}, parParams={'linewidth': 1.0, 'markersize': 5, 'markevery': 5, 'zorder': 3}, xlabelParams={'size': 10, 'xlabel': 'X'}, ylabelParams={'size': 10, 'ylabel': 'Y'}, xticksParams={'fontsize': 8, 'rotation': 0}, yticksParams={'fontsize': 8, 'rotation': 0}, legendParams={'fontsize': 8, 'frameon': False, 'loc': 'upper right', 'ncol': 2}, titleParams={'fontsize': 10, 'label': '@{frame} (${numberOfRemovedAtoms:.1f}$ $rem.$ $at.$) $Std.Err.={standardError:.3f}$\n$scale$ $factor$=${scaleFactor:.2f}$ - $multiframe$ $weight$=${multiframeWeight:.3f}$ - $used$ $({used})$'}, gridParams=None, show=True, **paramsKwargs)Plot constraint data. This can be overloaded in children classes.
| Parameters: |
|
|---|---|
| Returns: |
|
plot_multiframe_weights(frame, ax=None, titleFormat='@{frame} [subframes probability distribution]', show=True)plot multiframe subframes weight distribution histogram
| Parameters: |
|
|---|---|
| Returns: |
|
export(fileName, frame=None, format='%12.5f', delimiter='\t', comments='#')Export constraint data to text file or to an archive of files.
| Parameters: |
|
|---|
fullrmc.Core.Constraint.SingularConstraintBases: fullrmc.Core.Constraint.Constraint
A singular constraint is a constraint that doesn’t allow multiple instances in the same engine.
is_singular(engine)Get whether only one instance of this constraint type is present in the stochastic engine. True for only itself found, False for other instance of the same __class__.__name__ or constraintId.
| Parameters: |
|
|---|---|
| Returns: |
|
assert_singular(engine)Checks whether only one instance of this constraint type is present in the stochastic engine. Raises Exception if multiple instances are present.
fullrmc.Core.Constraint.RigidConstraint(rejectProbability)Bases: fullrmc.Core.Constraint.Constraint
A rigid constraint is a constraint that doesn’t count into the total standard error of the stochastic Engine. But it’s internal standard error must monotonously decrease or remain the same from one engine step to another. If standard error of an rigid constraint increases the step will be rejected even before engine’s new standardError get computed.
| Parameters: |
|
|---|
rejectProbabilityRejection probability.
set_reject_probability(rejectProbability)Set the rejection probability. This method doesn’t allow specifying frames. It will target used frame only.
| Parameters: |
|
|---|
should_step_get_rejected(standardError)Given a standard error, return whether to keep or reject new standard error according to the constraint reject probability.
| Parameters: | #. standardError (number): The standard error to compare with the Constraint standard error |
|---|---|
| Return: |
|
should_step_get_accepted(standardError)Given a standard error, return whether to keep or reject new standard error according to the constraint reject probability.
| Parameters: |
|
|---|---|
| Return: |
|
fullrmc.Core.Constraint.randfloat()random() -> x in the interval [0, 1).
Collection of methods and classes definition useful for constraints computation
fullrmc.Constraints.Collection.ShapeFunction(engine, weighting='atomicNumber', qmin=0.001, qmax=1, dq=0.005, rmin=0.0, rmax=100, dr=1)¶Bases: object
Shape function used to correct for particle shape. The shape function is subtracted from the total G(r) of g(r). It must be used when non-periodic boundary conditions are used to take into account the atomic density drop and to correct for the \(\rho_{0}\) approximation.
| Parameters: |
|
|---|
N.B: tweak qmax as small as possible to reduce the wriggles …
get_Gr_shape_function(rValues, compute=True)¶Get shape function of G(r) used in a PairDistributionConstraint.
| Parameters: |
|
|---|---|
| Returns: |
|
get_gr_shape_function(rValues, compute=True)¶Get shape function of g(r) used in a PairCorrelationConstraint.
| Parameters: |
|
|---|---|
| Returns: |
|
DistanceConstraints contains classes for all constraints related to distances between atoms.

fullrmc.Constraints.DistanceConstraints.InterMolecularDistanceConstraint(defaultDistance=1.5, typeDefinition='element', pairsDistanceDefinition=None, flexible=True, rejectProbability=1)¶Bases: fullrmc.Constraints.DistanceConstraints._MolecularDistanceConstraint
Its controls the inter-molecular distances between atoms.
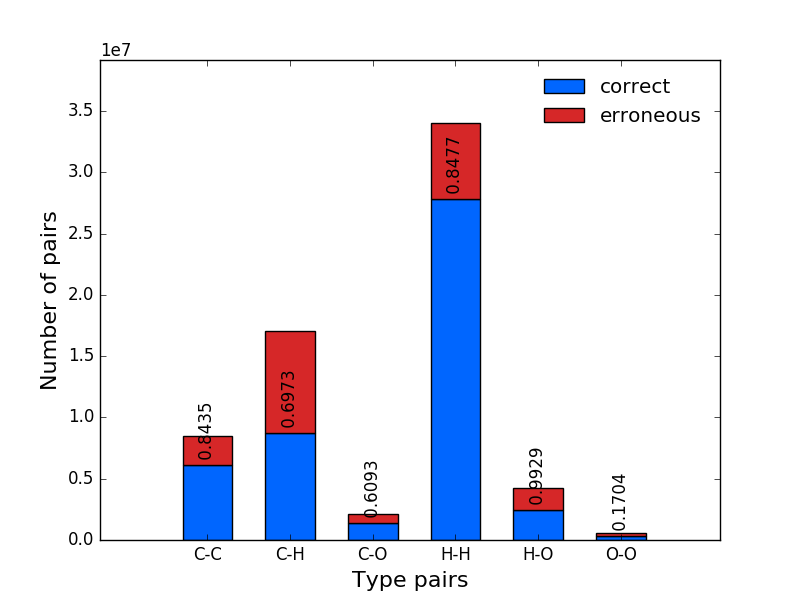
|
| Parameters: |
|
|---|
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Constraints.DistanceConstraints import InterMolecularDistanceConstraint
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# create and add constraint
EMD = InterMolecularDistanceConstraint()
ENGINE.add_constraints(EMD)
# create definition
EMD.set_pairs_distance([('Si','Si',1.75), ('O','O',1.10), ('Si','O',1.30)])
defaultDistance¶Default minimum distance.
pairsDistanceDefinition¶Pairs distance definition.
pairsDistance¶Pairs distance dictionary.
flexible¶Flexible flag.
lowerLimitArray¶Lower limit array used in distances calculation. for InterMolecularDistanceConstraint it’s always a numpy.zeros array
upperLimitArray¶Upper limit array used in distances calculation. for InterMolecularDistanceConstraint it’s the minimum distance allowed between pair of intermolecular atoms.
typePairs¶Atom’s type pairs sorted list.
typeDefinition¶Atom’s type definition.
types¶Atom’s type set.
allTypes¶All atoms type.
numberOfTypes¶Number of defined atom types in the configuration.
typesIndex¶Type indexes list.
numberOfAtomsPerType¶Number of atoms per type dict.
listen(message, argument=None)¶listen to any message sent from the Broadcaster.
| Parameters: |
|
|---|
set_flexible(flexible)¶Set flexible flag.
| Parameters: |
|
|---|
set_default_distance(defaultDistance)¶Sets the default intermolecular minimum distance.
| Parameters: |
|
|---|
set_type_definition(typeDefinition, pairsDistanceDefinition=None)¶Alias to set_pairs_distance used when typeDefinition needs to be re-defined.
| Parameters: |
|
|---|
set_pairs_distance(pairsDistanceDefinition)¶Set the pairs intermolecular minimum distance.
| Parameters: |
|
|---|
should_step_get_rejected(standardError)¶Given a standardError, return whether to keep or reject new standardError according to the constraint rejectProbability. In addition, if flexible flag is set to True, total number of atoms not satisfying constraints definition must be decreasing or at least remain the same.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_standard_error(data)¶Compute the standard error (stdErr) of data not satisfying constraint’s conditions.
Where:
\(N\) is the total number of atoms in the system.
\(D_{ij}\) is the distance constraint set for atoms pair (i,j).
\(d_{ij}\) is the distance between atom i and atom j.
\(\delta\) is the Dirac delta function.
\(\int_{0}^{D_{ij}} \delta(x-d_{ij}) dx\) is equal to 1 if \(0 \leqslant d_{ij} \leqslant D_{ij}\) and 0 elsewhere.
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraint_value()¶Get constraint’s formatted dictionary data.
| Returns: |
|
|---|
compute_data(*args, **kwargs)¶Compute constraint’s data.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_before_move(realIndexes, relativeIndexes)¶Compute constraint before move is executed
| Parameters: |
|
|---|
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Compute constraint’s data after move is executed.
| Parameters: |
|
|---|
accept_move(realIndexes, relativeIndexes)¶Accept move.
| Parameters: |
|
|---|
reject_move(realIndexes, relativeIndexes)¶Reject move.
| Parameters: |
|
|---|
compute_as_if_amputated(realIndex, relativeIndex)¶Compute and return constraint’s data and standard error as if atom given its its was amputated.
| Parameters: |
|
|---|
accept_amputation(realIndex, relativeIndex)¶Accept amputation of atom and sets constraints data and standard error accordingly.
| Parameters: |
|
|---|
reject_amputation(realIndex, relativeIndex)¶Reject amputation of atom.
| Parameters: |
|
|---|
plot(inBarParams={'color': '#0066ff', 'label': 'inbound', 'width': 0.6}, outBarParams={'color': '#d62728', 'label': 'outbound', 'width': 0.6}, txtParams={'color': 'black', 'fontsize': 8, 'horizontalalignment': 'center', 'rotation': 90, 'verticalalignment': 'center'}, xlabelParams={'size': 10, 'xlabel': 'Type pairs'}, ylabelParams={'size': 10, 'ylabel': 'Number of pairs'}, xticksParams={'fontsize': 8, 'rotation': 45}, **kwargs)¶Alias to Constraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|
export(fileName, frame=None, format='%s', delimiter='\t', comments='#')¶Export constraint data to text file or to an archive of files.
| Parameters: |
|
|---|
fullrmc.Constraints.DistanceConstraints.IntraMolecularDistanceConstraint(defaultDistance=1.5, typeDefinition='name', pairsDistanceDefinition=None, flexible=True, rejectProbability=1)¶Bases: fullrmc.Constraints.DistanceConstraints._MolecularDistanceConstraint
Its controls the intra-molecular distances between atoms.
|
|
| Parameters: |
|
|---|
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Constraints.DistanceConstraints import IntraMolecularDistanceConstraint
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# create and add constraint
EMD = IntraMolecularDistanceConstraint()
ENGINE.add_constraints(EMD)
# create definition
EMD.set_pairs_distance([('Si','Si',1.75), ('O','O',1.10), ('Si','O',1.30)])
defaultDistance¶Default minimum distance.
pairsDistanceDefinition¶Pairs distance definition.
pairsDistance¶Pairs distance dictionary.
flexible¶Flexible flag.
lowerLimitArray¶Lower limit array used in distances calculation. for InterMolecularDistanceConstraint it’s always a numpy.zeros array
upperLimitArray¶Upper limit array used in distances calculation. for InterMolecularDistanceConstraint it’s the minimum distance allowed between pair of intermolecular atoms.
typePairs¶Atom’s type pairs sorted list.
typeDefinition¶Atom’s type definition.
types¶Atom’s type set.
allTypes¶All atoms type.
numberOfTypes¶Number of defined atom types in the configuration.
typesIndex¶Type indexes list.
numberOfAtomsPerType¶Number of atoms per type dict.
listen(message, argument=None)¶listen to any message sent from the Broadcaster.
| Parameters: |
|
|---|
set_flexible(flexible)¶Set flexible flag.
| Parameters: |
|
|---|
set_default_distance(defaultDistance)¶Sets the default intermolecular minimum distance.
| Parameters: |
|
|---|
set_type_definition(typeDefinition, pairsDistanceDefinition=None)¶Alias to set_pairs_distance used when typeDefinition needs to be re-defined.
| Parameters: |
|
|---|
set_pairs_distance(pairsDistanceDefinition)¶Set the pairs intermolecular minimum distance.
| Parameters: |
|
|---|
should_step_get_rejected(standardError)¶Given a standardError, return whether to keep or reject new standardError according to the constraint rejectProbability. In addition, if flexible flag is set to True, total number of atoms not satisfying constraints definition must be decreasing or at least remain the same.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_standard_error(data)¶Compute the standard error (stdErr) of data not satisfying constraint’s conditions.
Where:
\(N\) is the total number of atoms in the system.
\(D_{ij}\) is the distance constraint set for atoms pair (i,j).
\(d_{ij}\) is the distance between atom i and atom j.
\(\delta\) is the Dirac delta function.
\(\int_{0}^{D_{ij}} \delta(x-d_{ij}) dx\) is equal to 1 if \(0 \leqslant d_{ij} \leqslant D_{ij}\) and 0 elsewhere.
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraint_value()¶Get constraint’s formatted dictionary data.
| Returns: |
|
|---|
compute_data(*args, **kwargs)¶Compute constraint’s data.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_before_move(realIndexes, relativeIndexes)¶Compute constraint before move is executed
| Parameters: |
|
|---|
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Compute constraint’s data after move is executed.
| Parameters: |
|
|---|
accept_move(realIndexes, relativeIndexes)¶Accept move.
| Parameters: |
|
|---|
reject_move(realIndexes, relativeIndexes)¶Reject move.
| Parameters: |
|
|---|
compute_as_if_amputated(realIndex, relativeIndex)¶Compute and return constraint’s data and standard error as if atom given its its was amputated.
| Parameters: |
|
|---|
accept_amputation(realIndex, relativeIndex)¶Accept amputation of atom and sets constraints data and standard error accordingly.
| Parameters: |
|
|---|
reject_amputation(realIndex, relativeIndex)¶Reject amputation of atom.
| Parameters: |
|
|---|
plot(inBarParams={'color': '#0066ff', 'label': 'inbound', 'width': 0.6}, outBarParams={'color': '#d62728', 'label': 'outbound', 'width': 0.6}, txtParams={'color': 'black', 'fontsize': 8, 'horizontalalignment': 'center', 'rotation': 90, 'verticalalignment': 'center'}, xlabelParams={'size': 10, 'xlabel': 'Type pairs'}, ylabelParams={'size': 10, 'ylabel': 'Number of pairs'}, xticksParams={'fontsize': 8, 'rotation': 45}, **kwargs)¶Alias to Constraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|
export(fileName, frame=None, format='%s', delimiter='\t', comments='#')¶Export constraint data to text file or to an archive of files.
| Parameters: |
|
|---|
AtomicCoordinationConstraints contains classes for all constraints related to coordination number in spherical shells around atoms.

fullrmc.Constraints.AtomicCoordinationConstraints.AtomicCoordinationNumberConstraint(rejectProbability=1)¶Bases: fullrmc.Core.Constraint.RigidConstraint, fullrmc.Core.Constraint.SingularConstraint
It’s a rigid constraint that controls the coordination number of atoms.
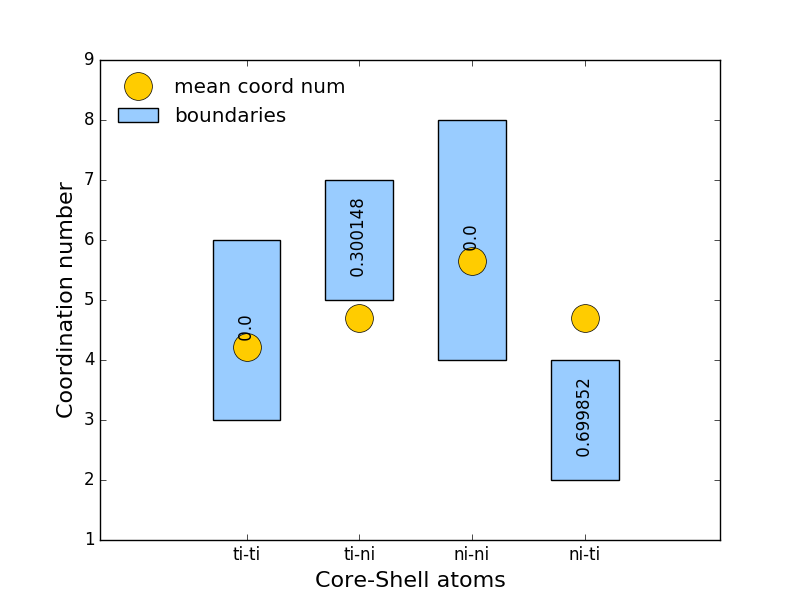
|
| Parameters: |
|
|---|
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Constraints.AtomicCoordinationConstraints import AtomicCoordinationNumberConstraint
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# create and add constraint
ACNC = AtomicCoordinationNumberConstraint()
ENGINE.add_constraints(ACNC)
# create definition
ACNC.set_coordination_number_definition( [ ('Al','Cl',1.5, 2.5, 2, 2),
('Al','S', 2.5, 3.0, 2, 2)] )
coordNumDef¶Copy of coordination number definition dictionary
coordinationNumberDefinition¶alias to coordinationNumberDefinition
coresIndexes¶List of coordination number core atoms index array.
shellsIndexes¶List of coordination number shell atoms index array.
numberOfCores¶Array of number of core atoms
lowerShells¶Array of lower shells distance.
upperShells¶Array of upper shells distance.
minAtoms¶Array of minimum number of atoms in a shell.
maxAtoms¶Array of maximum number of atoms in a shell.
weights¶Shells weight which count in the computation of standard error.
data¶Coordination number constraint data.
asCoreDefIdxs¶List of arrays where each element is pointing to a coordination number definition where the atom is a core.
inShellDefIdxs¶List of arrays where each element is pointing to a coordination number definition where the atom is in a shell.
listen(message, argument=None)¶listen to any message sent from the Broadcaster.
| Parameters: |
|
|---|
set_coordination_number_definition(coordNumDef)¶Set the coordination number definition.
| Parameters: |
|
|---|
compute_standard_error(data)¶Compute the standard error (StdErr) of data not satisfying constraint’s conditions.
Where:
\(S\) is the total number of defined coordination number shells.
\(W_{i}\) is the defined weight of coordination number shell i.
\(Dev_{i}\) is the standard deviation of the coordination number in shell definition i.
\(\overline{CN_{i}}\) is the mean coordination number value in shell definition i.
\(N_{min,i}\) is the defined minimum number of neighbours in shell definition i.
\(N_{max,i}\) is the defined maximum number of neighbours in shell definition i.
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraint_value()¶Get constraint’s data.
| Returns: |
|
|---|
compute_data(*args, **kwargs)¶Compute constraint’s data.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_before_move(realIndexes, relativeIndexes)¶Compute constraint’s data before move is executed.
| Parameters: |
|
|---|
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Compute constraint’s data after move is executed.
| Parameters: |
|
|---|
accept_move(realIndexes, relativeIndexes)¶Accept move.
| Parameters: |
|
|---|
reject_move(realIndexes, relativeIndexes)¶Reject move.
| Parameters: |
|
|---|
compute_as_if_amputated(realIndex, relativeIndex)¶Compute and return constraint’s data and standard error as if atom given its its was amputated.
| Parameters: |
|
|---|
accept_amputation(realIndex, relativeIndex)¶Accept amputation of atom and sets constraints data and standard error accordingly.
| Parameters: |
|
|---|
reject_amputation(realIndex, relativeIndex)¶Reject amputation of atom.
| Parameters: |
|
|---|
plot(dataParams={'color': '#ffcc00', 'label': 'mean coord num', 'linewidth': 0, 'marker': 'o', 'markersize': 20, 'markevery': 1}, barParams={'color': '#99ccff', 'label': 'boundaries', 'width': 0.6}, txtParams={'color': 'black', 'fontsize': 8, 'horizontalalignment': 'center', 'rotation': 90, 'verticalalignment': 'center'}, xlabelParams={'size': 10, 'xlabel': 'Core-Shell atoms'}, ylabelParams={'size': 10, 'ylabel': 'Coordination number'}, xticksParams={'fontsize': 8, 'rotation': 45}, titleParams={'fontsize': 8, 'label': '@{frame} (${numberOfRemovedAtoms:.1f}$ $rem.$ $at.$) $Std.Err.={standardError:.3f}$'}, **kwargs)¶Alias to Constraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|
BondConstraints contains classes for all constraints related to bond length between atoms.
fullrmc.Constraints.BondConstraints.BondConstraint(rejectProbability=1)¶Bases: fullrmc.Core.Constraint.RigidConstraint, fullrmc.Core.Constraint.SingularConstraint
Controls the bond’s length defined between two atoms.
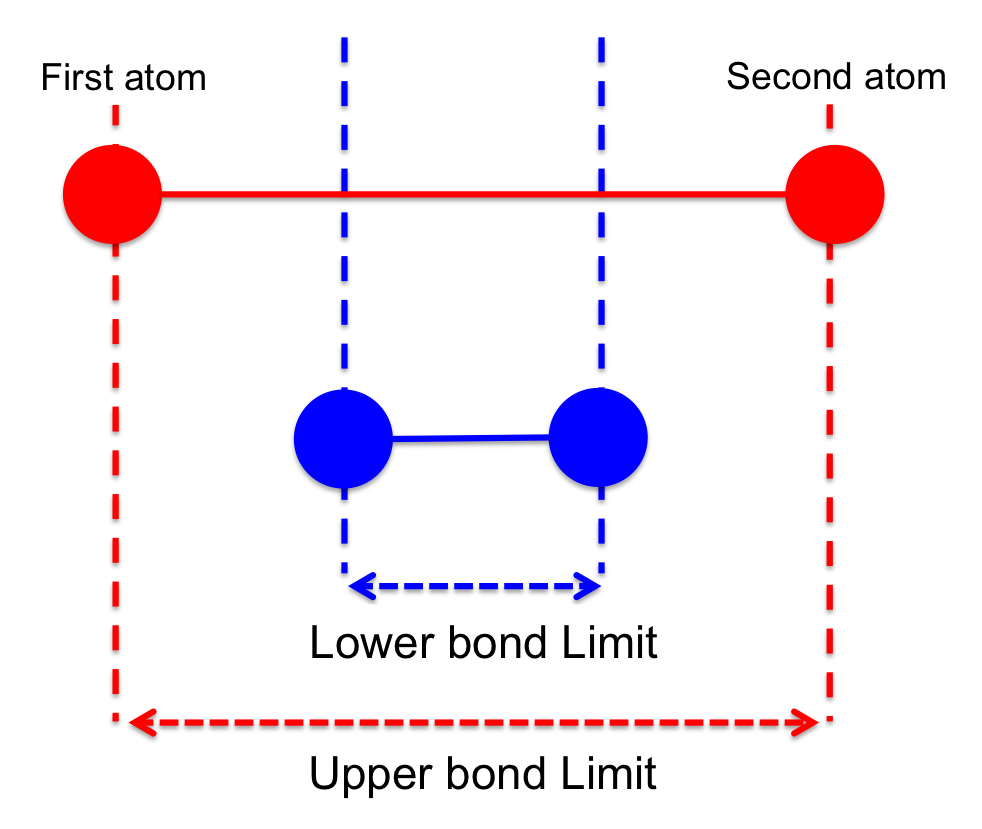
Bond sketch defined between two atoms. |
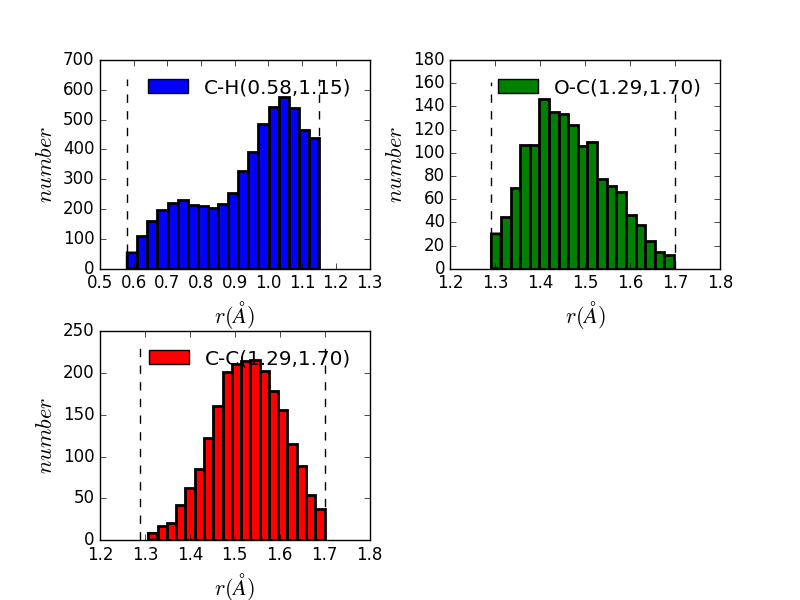
|
| Parameters: |
|
|---|
## Water (H2O) molecule sketch
##
## O
## / \
## / H2O \
## H1 H2
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Constraints.BondConstraints import BondConstraint
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# create and add constraint
BC = BondConstraint()
ENGINE.add_constraints(BC)
# define intra-molecular bonds
BC.create_bonds_by_definition( bondsDefinition={"H2O": [ ('O','H1', 0.88, 1.02),
('O','H2', 0.88, 1.02) ]} )
bondsList¶List of defined bonds
bondsDefinition¶bonds definition copy if bonds are defined as such
bonds¶Bonds dictionary map of every and each atom
listen(message, argument=None)¶Listens to any message sent from the Broadcaster.
| Parameters: |
|
|---|
set_bonds(bondsList, tform=True)¶Sets bonds dictionary by parsing bondsList list.
| Parameters: |
|
|---|
create_bonds_by_definition(bondsDefinition)¶Creates bondsList using bonds definition. Calls set_bonds(bondsList) and generates bonds attribute.
| Parameters: |
e.g. (Carbon tetrachloride): bondsDefinition={"CCL4": [('C','CL1' , 1.55, 1.95),
('C','CL2' , 1.55, 1.95),
('C','CL3' , 1.55, 1.95),
('C','CL4' , 1.55, 1.95) ] }
|
|---|
compute_standard_error(data)¶Compute the standard error (StdErr) of data not satisfying constraint conditions.
Where:
\(C\) is the total number of defined bonds constraints.
\(\beta_{i}^{min}\) is the bond constraint lower limit set for constraint i.
\(\beta_{i}^{max}\) is the bond constraint upper limit set for constraint i.
\(\beta_{i}\) is the bond length computed for constraint i.
\(\delta\) is the Dirac delta function.
\(\int_{0}^{\beta_{i}^{min}} \delta(\beta-\beta_{i}) d \beta\) is equal to 1 if \(0 \leqslant \beta_{i} \leqslant \beta_{i}^{min}\) and 0 elsewhere.
\(\int_{\beta_{i}^{max}}^{\pi} \delta(\beta-\beta_{i}) d \beta\) is equal to 1 if \(\beta_{i}^{max} \leqslant \beta_{i} \leqslant \infty\) and 0 elsewhere.
| Parameters: |
|
|---|---|
| Returns: | #. standardError (number): The calculated standardError of the given. |
get_constraint_value()¶Get constraint’s data.
| Returns: |
|
|---|
compute_data(*args, **kwargs)¶Compute constraint’s data.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_before_move(realIndexes, relativeIndexes)¶Compute constraint’s data before move is executed.
| Parameters: |
|
|---|
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Compute constraint after move is executed
| Parameters: |
|
|---|
accept_move(realIndexes, relativeIndexes)¶Accept move
| Parameters: |
|
|---|
reject_move(realIndexes, relativeIndexes)¶Reject move
| Parameters: |
|
|---|
compute_as_if_amputated(realIndex, relativeIndex)¶Compute and return constraint’s data and standard error as if atom given its its was amputated.
| Parameters: |
|
|---|
accept_amputation(realIndex, relativeIndex)¶Accept amputation of atom and sets constraints data and standard error accordingly.
| Parameters: |
|
|---|
reject_amputation(realIndex, relativeIndex)¶Reject amputation of atom.
| Parameters: |
|
|---|
plot(spacing=0.1, numberOfTicks=3, nbins=20, barsRelativeWidth=0.95, splitBy=None, stackHorizontal=True, colorCodeXticksLabels=True, xlabelParams={'size': 10, 'xlabel': '$r(\\AA)$'}, ylabelParams={'size': 10, 'ylabel': 'number'}, limitsParams={'color': None, 'linestyle': '--', 'linewidth': 1.0}, **kwargs)¶Alias to Constraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|
AngleConstraints contains classes for all constraints related angles between atoms.
fullrmc.Constraints.AngleConstraints.BondsAngleConstraint(rejectProbability=1)¶Bases: fullrmc.Core.Constraint.RigidConstraint, fullrmc.Core.Constraint.SingularConstraint
Controls angle defined between 3 defined atoms, a first atom called central and the remain two called left and right.
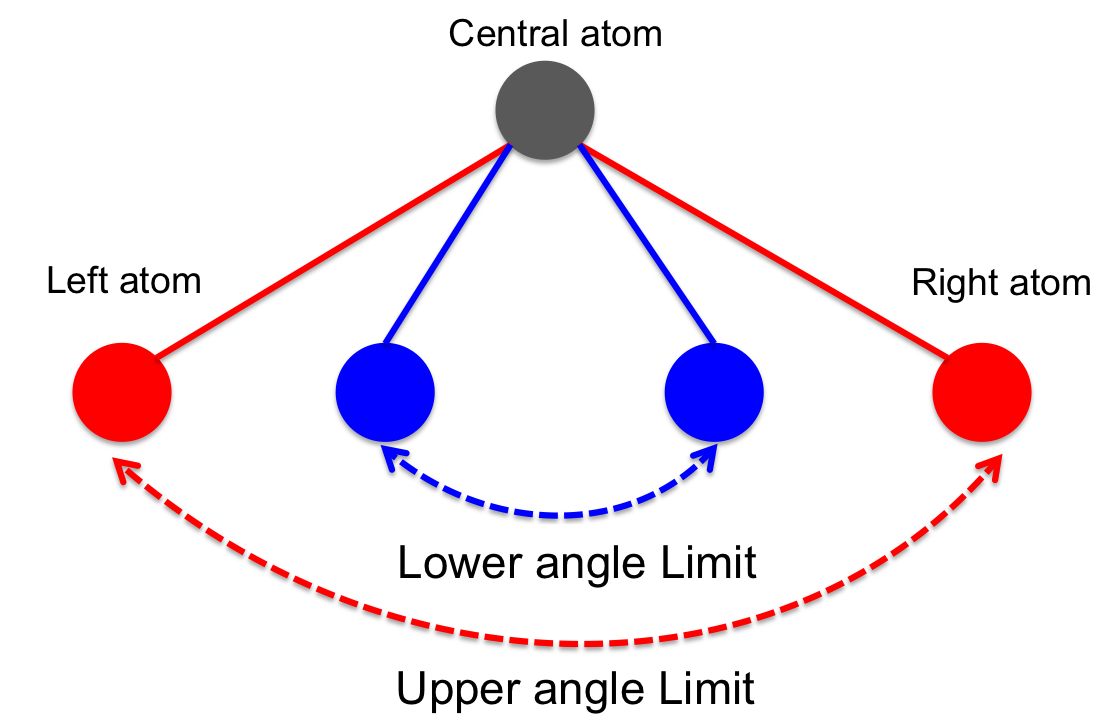
Angle sketch defined between three atoms. |
|
| Parameters: |
|
|---|
## Methane (CH4) molecule sketch
##
## H4
## |
## |
## _- C -_
## H1- / -_
## / H3
## H2
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Constraints.AngleConstraints import BondsAngleConstraint
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# create and add constraint
BAC = BondsAngleConstraint()
ENGINE.add_constraints(BAC)
# define intra-molecular angles
BAC.create_angles_by_definition( anglesDefinition={"CH4": [ ('C','H1','H2', 100, 120),
('C','H2','H3', 100, 120),
('C','H3','H4', 100, 120),
('C','H4','H1', 100, 120) ]} )
anglesList¶Defined angles list.
anglesDefinition¶angles definition copy if angles are defined as such
angles¶Angles dictionary of every and each atom.
listen(message, argument=None)¶Listen to any message sent from the Broadcaster.
| Parameters: |
|
|---|
set_angles(anglesList, tform=True)¶Sets the angles dictionary by parsing the anglesList. All angles are in degrees.
| Parameters: |
|
|---|
create_angles_by_definition(anglesDefinition)¶Creates anglesList using angles definition. Calls set_angles(anglesList) and generates angles attribute.
| Parameters: |
|
|---|
e.g. (Carbon tetrachloride): anglesDefinition={"CCL4": [('C','CL1','CL2' , 105, 115),
('C','CL2','CL3' , 105, 115),
('C','CL3','CL4' , 105, 115),
('C','CL4','CL1' , 105, 115) ] }
compute_standard_error(data)¶Compute the standard error (StdErr) of data not satisfying constraint conditions.
Where:
\(C\) is the total number of defined angles constraints.
\(\theta_{i}^{min}\) is the angle constraint lower limit set for constraint i.
\(\theta_{i}^{max}\) is the angle constraint upper limit set for constraint i.
\(\theta_{i}\) is the angle computed for constraint i.
\(\delta\) is the Dirac delta function.
\(\int_{0}^{\theta_{i}^{min}} \delta(\theta-\theta_{i}) d \theta\) is equal to 1 if \(0 \leqslant \theta_{i} \leqslant \theta_{i}^{min}\) and 0 elsewhere.
\(\int_{\theta_{i}^{max}}^{\pi} \delta(\theta-\theta_{i}) d \theta\) is equal to 1 if \(\theta_{i}^{max} \leqslant \theta_{i} \leqslant \pi\) and 0 elsewhere.
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraint_value()¶Get partial Mean Pair Distances (MPD) below the defined minimum distance.
| Returns: |
|
|---|
compute_data(*args, **kwargs)¶Compute constraint’s data.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_before_move(realIndexes, relativeIndexes)¶Compute constraint before move is executed.
| Parameters: |
|
|---|
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Compute constraint after move is executed.
| Parameters: |
|
|---|
accept_move(realIndexes, relativeIndexes)¶Accept move.
| Parameters: |
|
|---|
reject_move(realIndexes, relativeIndexes)¶Reject move.
| Parameters: |
|
|---|
accept_amputation(realIndex, relativeIndex)¶Accept amputation of atom and set constraint’s data and standard error accordingly.
| Parameters: |
|
|---|
reject_amputation(realIndex, relativeIndex)¶Reject amputation of atom.
| Parameters: |
|
|---|
plot(spacing=2, numberOfTicks=2, nbins=20, barsRelativeWidth=0.95, splitBy=None, stackHorizontal=True, colorCodeXticksLabels=True, xlabelParams={'size': 10, 'xlabel': '$deg.$'}, ylabelParams={'size': 10, 'ylabel': 'number'}, limitsParams={'color': None, 'linestyle': '--', 'linewidth': 1.0}, **kwargs)¶Alias to Constraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|
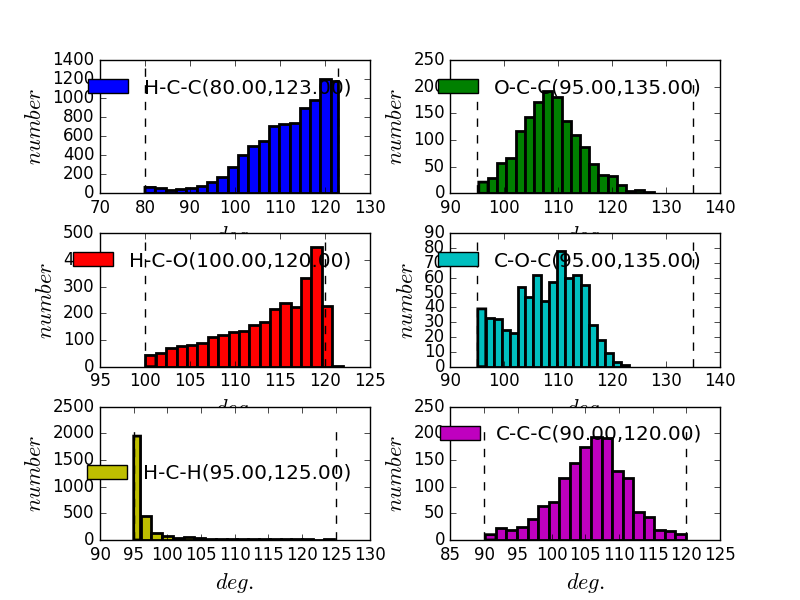
ImproperAngleConstraints contains classes for all constraint’s related to improper angles between bonded atoms.
fullrmc.Constraints.DihedralAngleConstraints.DihedralAngleConstraint(rejectProbability=1)¶Bases: fullrmc.Core.Constraint.RigidConstraint, fullrmc.Core.Constraint.SingularConstraint
Dihedral angle is defined between two intersecting planes formed with defined atoms. Dihedral angle constraint can control up to three angle shells at the same times.
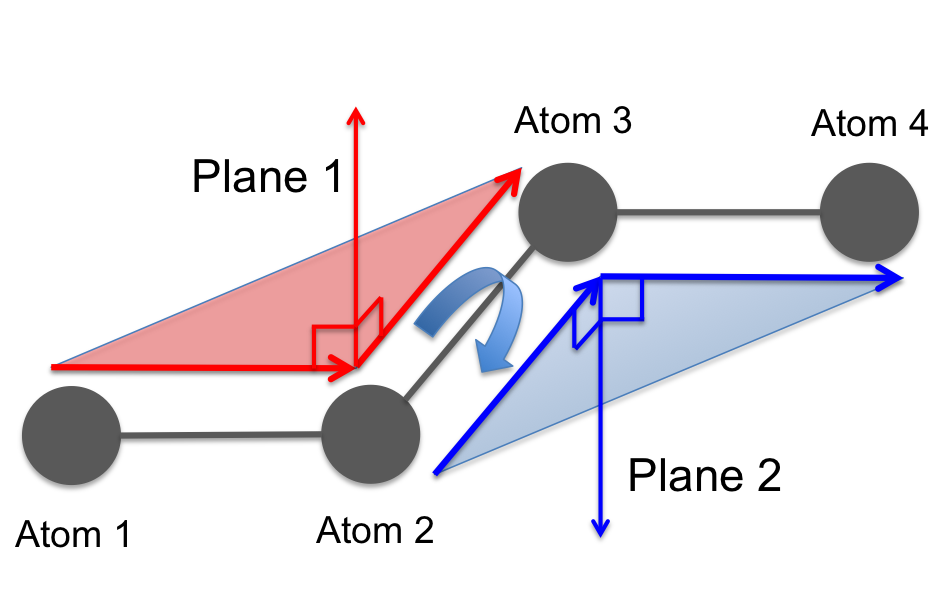
Dihedral angle sketch defined between two planes formed with four atoms. |
| Parameters: |
|
|---|
## Butane (BUT) molecule sketch
##
## H13 H22 H32 H43
## | | | |
## H11---C1---C2---C3---C4---H41
## | | | |
## H12 H21 H31 H42
##
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Constraints.DihedralAngleConstraints import DihedralAngleConstraint
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# create and add constraint
DAC = DihedralAngleConstraint()
ENGINE.add_constraints(DAC)
# define intra-molecular dihedral angles
DAC.create_angles_by_definition( anglesDefinition={"BUT": [ ('C1','C2','C3','C4', 40,80, 100,140, 290,330), ] })
anglesList¶Improper angles list.
anglesDefinition¶angles definition copy if dihedral angles are defined as such
angles¶Angles dictionary for every and each atom.
listen(message, argument=None)¶Listens to any message sent from the Broadcaster.
| Parameters: |
|
|---|
set_angles(anglesList, tform=True)¶Sets the angles dictionary by parsing the anglesList list. All angles are in degrees. Dihedral angle can control up to three angle shells at the same times defined using three different lower and upper angle bounds simulating three different dihedral potential energy minimums. Dihedral angles are defined from 0 to 360 degrees. Shell’s lower and upper bound defines a dihedral angle clockwise. Therefore in order to take into considerations the limits at 0 and 360 degrees, lower bound is allowed to be higher than the higher bound.
e.g. (50, 100) is a dihedral shell defined in the angle range between 50 and 100 degrees. But (100, 50) dihedral shell is defined between 100 to 360 degrees and wraps the range from 0 to 100. (50, 100) and (100, 50) are complementary and cover the whole range from 0 to 360 deg.
| Parameters: |
|
|---|
N.B. Defining three shells boundaries is mandatory. In case fewer than three shells is needed, it suffices to repeat one of the shells boundaries.
e.g. (‘C1’,’C2’,’C3’,’C4’, 40,80, 100,140, 40,80), in the herein definition the last shell is a repetition of the first which means only two shells are defined.
create_angles_by_definition(anglesDefinition)¶Creates anglesList using angles definition. Calls set_angles(anglesMap) and generates angles attribute.
| Parameters: |
|
|---|
e.g. (Butane): anglesDefinition={"BUT": [ ('C1','C2','C3','C4', 40,80, 100,140, 290,330), ] }
compute_standard_error(data)¶Compute the standard error (StdErr) of data not satisfying constraint conditions.
Where:
\(C\) is the total number of defined improper angles constraints.
\(\theta_{i}^{min}\) is the improper angle constraint lower limit set for constraint i.
\(\theta_{i}^{max}\) is the improper angle constraint upper limit set for constraint i.
\(\theta_{i}\) is the improper angle computed for constraint i.
\(\delta\) is the Dirac delta function.
\(\int_{0}^{\theta_{i}^{min}} \delta(\theta-\theta_{i}) d \theta\) is equal to 1 if \(0 \leqslant \theta_{i} \leqslant \theta_{i}^{min}\) and 0 elsewhere.
\(\int_{\theta_{i}^{max}}^{\pi} \delta(\theta-\theta_{i}) d \theta\) is equal to 1 if \(\theta_{i}^{max} \leqslant \theta_{i} \leqslant \pi\) and 0 elsewhere.
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraint_value()¶Get constraint’s data.
| Returns: |
|
|---|
compute_data(*args, **kwargs)¶Compute constraint’s data.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_before_move(realIndexes, relativeIndexes)¶Compute constraint’s data before move is executed.
| Parameters: |
|
|---|
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Compute constraint’s data after move is executed.
| Parameters: |
|
|---|
accept_move(realIndexes, relativeIndexes)¶Accept move.
reject_move(realIndexes, relativeIndexes)¶Reject move
| Parameters: |
|
|---|
accept_amputation(realIndex, relativeIndex)¶Accept amputation of atom and set constraint’s data and standard error accordingly.
| Parameters: |
|
|---|
reject_amputation(realIndex, relativeIndex)¶Reject amputation of atom.
| Parameters: |
|
|---|
plot(spacing=2, numberOfTicks=3, nbins=20, barsRelativeWidth=0.95, splitBy=None, stackHorizontal=False, colorCodeXticksLabels=True, xlabelParams={'size': 10, 'xlabel': '$deg.$'}, ylabelParams={'size': 10, 'ylabel': 'number'}, limitsParams={'color': None, 'linestyle': None, 'linewidth': 1.0}, **kwargs)¶Alias to Constraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|
ImproperAngleConstraints contains classes for all constraints related to improper angles between atoms.
fullrmc.Constraints.ImproperAngleConstraints.ImproperAngleConstraint(rejectProbability=1)¶Bases: fullrmc.Core.Constraint.RigidConstraint, fullrmc.Core.Constraint.SingularConstraint
Controls the improper angle formed with 4 defined atoms. It’s mainly used to keep the improper atom in the plane defined with three other atoms. The improper vector is defined as the vector from the first atom of the plane to the improper atom. Therefore the improper angle is defined between the improper vector and the plane.
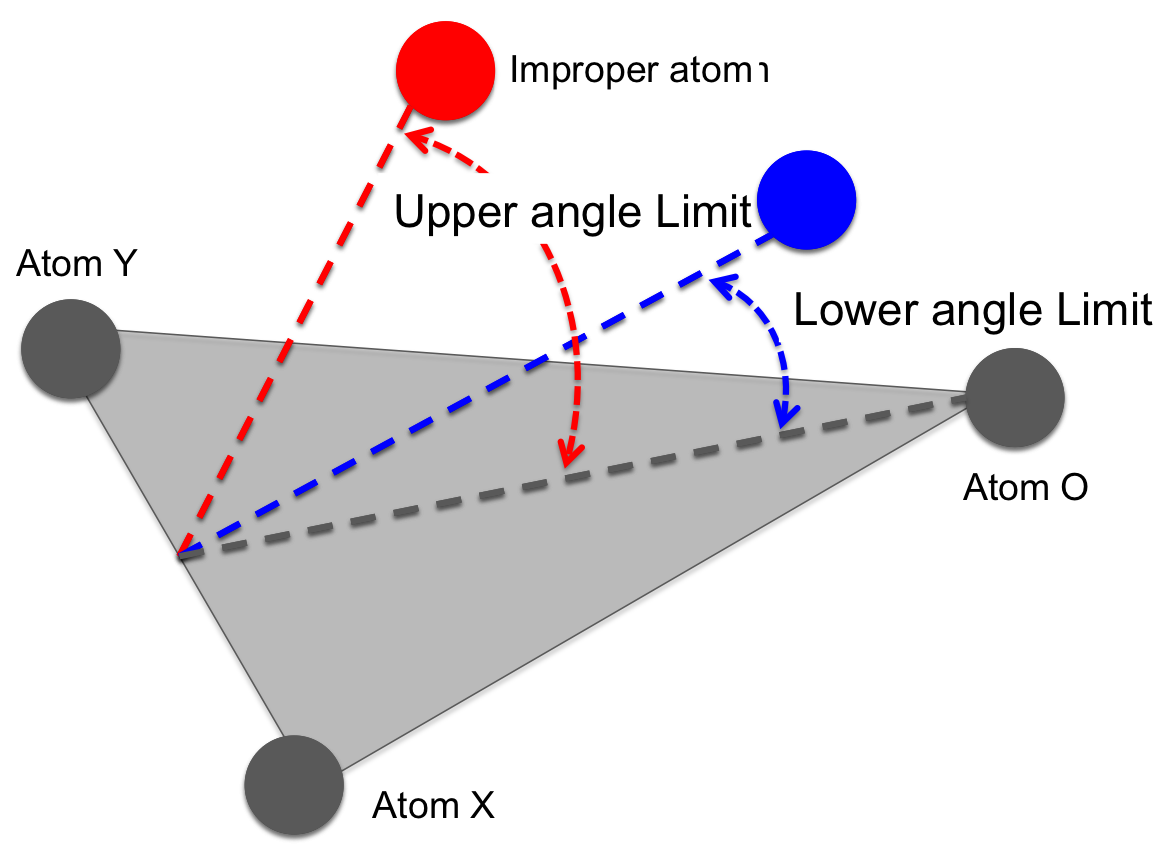
Improper angle sketch defined between four atoms. |
| Parameters: |
|
|---|
## Tetrahydrofuran (THF) molecule sketch
##
## O
## H41 / \ H11
## \ / \ /
## H42-- C4 THF C1 --H12
## \ MOLECULE /
## \ /
## H31-- C3-------C2 --H21
## / \
## H32 H22
##
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Constraints.ImproperAngleConstraints import ImproperAngleConstraint
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# create and add constraint
IAC = ImproperAngleConstraint()
ENGINE.add_constraints(IAC)
# define intra-molecular improper angles
IAC.create_angles_by_definition( anglesDefinition={"THF": [ ('C2','O','C1','C4', -15, 15),
('C3','O','C1','C4', -15, 15) ] })
anglesList¶Get improper angles list.
anglesDefinition¶angles definition copy if improper angles are defined as such
angles¶Get angles dictionary for every and each atom.
listen(message, argument=None)¶Listens to any message sent from the Broadcaster.
| Parameters: |
|
|---|
set_angles(anglesList, tform=True)¶Set angles dictionary by parsing anglesList list.
| Parameters: |
|
|---|
create_angles_by_definition(anglesDefinition)¶Creates anglesList using angles definition. This calls set_angles(anglesMap) and generates angles attribute. All angles are in degrees.
| Parameters: |
|
|---|
e.g. (Benzene): anglesDefinition={"BENZ": [('C3','C1','C2','C6', -10, 10),
('C4','C1','C2','C6', -10, 10),
('C5','C1','C2','C6', -10, 10) ] }
compute_standard_error(data)¶Compute the standard error (StdErr) of data not satisfying constraint’s conditions.
Where:
\(C\) is the total number of defined improper angles constraints.
\(\theta_{i}^{min}\) is the improper angle constraint lower limit set for constraint i.
\(\theta_{i}^{max}\) is the improper angle constraint upper limit set for constraint i.
\(\theta_{i}\) is the improper angle computed for constraint i.
\(\delta\) is the Dirac delta function.
\(\int_{0}^{\theta_{i}^{min}} \delta(\theta-\theta_{i}) d \theta\) is equal to 1 if \(0 \leqslant \theta_{i} \leqslant \theta_{i}^{min}\) and 0 elsewhere.
\(\int_{\theta_{i}^{max}}^{\pi} \delta(\theta-\theta_{i}) d \theta\) is equal to 1 if \(\theta_{i}^{max} \leqslant \theta_{i} \leqslant \pi\) and 0 elsewhere.
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraint_value()¶get constraint’s data value.
| Returns: |
|
|---|
compute_data(*args, **kwargs)¶Compute constraint’s data.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_before_move(realIndexes, relativeIndexes)¶Compute constraint’s data before move is executed.
| Parameters: |
|
|---|
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Compute constraint’s data after move is executed.
| Parameters: |
|
|---|
accept_move(realIndexes, relativeIndexes)¶Accept move
| Parameters: |
|
|---|
reject_move(realIndexes, relativeIndexes)¶Reject move
| Parameters: |
|
|---|
accept_amputation(realIndex, relativeIndex)¶Accept amputation of atom and sets constraint’s data and standard error accordingly.
| Parameters: |
|
|---|
reject_amputation(realIndex, relativeIndex)¶Reject amputation of atom.
| Parameters: |
|
|---|
plot(spacing=2, numberOfTicks=2, nbins=20, barsRelativeWidth=0.95, splitBy=None, stackHorizontal=True, colorCodeXticksLabels=True, xlabelParams={'size': 10, 'xlabel': '$deg.$'}, ylabelParams={'size': 10, 'ylabel': 'number'}, limitsParams={'color': None, 'linestyle': '--', 'linewidth': 1.0}, **kwargs)¶Alias to Constraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|
PairDistributionConstraints contains classes for all constraints related to experimental pair distribution functions.

fullrmc.Constraints.PairDistributionConstraints.PairDistributionConstraint(experimentalData, dataWeights=None, weighting='atomicNumber', atomsWeight=None, scaleFactor=1.0, adjustScaleFactor=(0, 0.8, 1.2), shapeFuncParams=None, windowFunction=None, limits=None)¶Bases: fullrmc.Core.Constraint.ExperimentalConstraint
Controls the total reduced pair distribution function (pdf) of atomic configuration noted as G(r). The pair distribution function is directly calculated from powder diffraction experimental data. It is obtained from the experimentally determined total-scattering structure function S(Q), by a Sine Fourier transform according to.
Theoretically G(r) oscillates about zero. Also \(G(r) \rightarrow 0\) when \(r \rightarrow \infty\) and \(G(r) \rightarrow 0\) when \(r \rightarrow 0\) with a slope of \(-4\pi\rho_{0}\) where \(\rho_{0}\) is the number density of the material.
Model wise, G(r) is computed after calculating the so called Pair Correlation Function noted as g(r). The relation between G(r) and g(r) is given by
\(\rho_{r}\) is the number density fluctuation at distance \(r\). The computation of g(r) is straightforward from an atomistic model and it is given by \(g(r)=\rho_{r} / \rho_{0}\).
The radial distribution function noted \(R(r)\) is a very important function because it describes directly the system’s structure. \(R(r)dr\) gives the number of atoms in an annulus of thickness dr at distance r from another atom. Therefore, the coordination number, or the number of neighbors within the distances interval \([a,b]\) is given by \(\int_{a}^{b} R(r) dr\)
Finally, g(r) is calculated after binning all pair atomic distances into a weighted histograms of values \(n(r)\) from which local number densities are computed as the following:
Where:
\(Q\) is the momentum transfer.
\(r\) is the distance between two atoms.
\(\rho_{i,j}(r)\) is the pair density function of atoms i and j.
\(\rho_{0}\) is the average number density of the system.
\(w_{i,j}\) is the relative weighting of atom types i and j.
\(R(r)\) is the radial distribution function (rdf).
\(N\) is the total number of atoms.
\(V\) is the volume of the system.
\(n_{i,j}(r)\) is the number of atoms i neighbouring j at a distance r.
\(v(r)\) is the annulus volume at distance r and of thickness dr.
\(N_{i,j}\) is the total number of atoms i and j in the system.
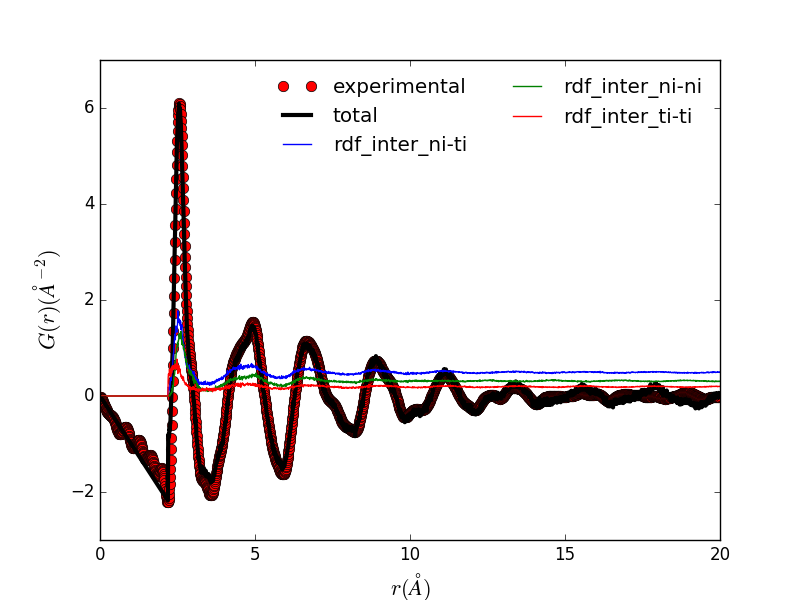
|
| Parameters: |
|
|---|
NB: If adjustScaleFactor first item (frequency) is 0, the scale factor will remain untouched and the limits minimum and maximum won’t be checked.
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Constraints.PairDistributionConstraints import PairDistributionConstraint
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# create and add constraint
PDC = PairDistributionConstraint(experimentalData="pdf.dat", weighting="atomicNumber")
ENGINE.add_constraints(PDC)
bin¶Experimental data distances bin.
minimumDistance¶Experimental data minimum distances.
maximumDistance¶Experimental data maximum distances.
histogramSize¶Histogram size.
experimentalDistances¶Experimental distances array.
shellCenters¶Shells center array.
shellVolumes¶Shells volume array.
experimentalPDF¶Experimental pair distribution function data.
elementsPairs¶Elements pairs.
weighting¶Elements weighting definition.
atomsWeight¶Custom atoms weight
weightingScheme¶Elements weighting scheme.
windowFunction¶Window function.
shapeArray¶Shape function data array.
shapeUpdateFreq¶Shape function update frequency.
listen(message, argument=None)¶Listens to any message sent from the Broadcaster.
| Parameters: |
|
|---|
set_shape_function_parameters(shapeFuncParams)¶Set the shape function. The shape function can be set to None which means unsused, or set as a constant shape given by a numpy.ndarray or computed from all atoms and updated every ‘updateFreq’ accepted moves. The shape function is subtracted from the total G(r). It must be used when non-periodic boundary conditions are used to take into account the atomic density drop and to correct for the \(\rho_{0}\) approximation.
| Parameters: |
|
|---|
set_weighting(weighting)¶Set elements weighting. It must be a valid entry of pdbparser atom’s database.
| Parameters: |
|
|---|
set_atoms_weight(atomsWeight)¶Custom set atoms weight. This is the way to customize setting atoms weights different than the given weighting scheme.
| Parameters: |
|
|---|
set_window_function(windowFunction, frame=None)¶Set convolution window function.
| Parameters: |
|
|---|
set_experimental_data(experimentalData)¶Set constraint’s experimental data.
| Parameters: |
|
|---|
set_limits(limits)¶Set the histogram computation limits.
| Parameters: |
|
|---|
check_experimental_data(experimentalData)¶Check whether experimental data is correct.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_standard_error(modelData)¶Compute the standard error (StdErr) as the squared deviations between model computed data and the experimental ones.
Where:
\(N\) is the total number of experimental data points.
\(W_{i}\) is the data point weight. It becomes equivalent to 1 when dataWeights is set to None.
\(Y(X_{i})\) is the experimental data point \(X_{i}\).
\(F(X_{i})\) is the computed from the model data \(X_{i}\).
| Parameters: |
|
|---|---|
| Returns: |
|
update_standard_error()¶Compute and set constraint’s standardError.
get_constraint_value(applyMultiframePrior=True)¶Compute all partial Pair Distribution Functions (PDFs).
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraint_original_value()¶Compute all partial Pair Distribution Functions (PDFs).
| Returns: |
|
|---|
compute_data(*args, **kwargs)¶Compute constraint’s data.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_before_move(realIndexes, relativeIndexes)¶Compute constraint before move is executed.
| Parameters: |
|
|---|
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Compute constraint after move is executed
| Parameters: |
|
|---|
accept_move(realIndexes, relativeIndexes)¶Accept move
| Parameters: |
|
|---|
reject_move(realIndexes, relativeIndexes)¶Reject move
| Parameters: |
|
|---|
compute_as_if_amputated(realIndex, relativeIndex)¶Compute and return constraint’s data and standard error as if given atom is amputated.
| Parameters: |
|
|---|
accept_amputation(realIndex, relativeIndex)¶Accept amputated atom and sets constraints data and standard error accordingly.
| Parameters: |
|
|---|
reject_amputation(realIndex, relativeIndex)¶Reject amputated atom and set constraint’s data and standard error accordingly.
| Parameters: |
|
|---|
get_multiframe_weights(frame)¶plot(xlabelParams={'size': 10, 'xlabel': '$r(\\AA)$'}, ylabelParams={'size': 10, 'ylabel': '$G(r)(\\AA^{-2})$'}, **kwargs)¶Alias to ExperimentalConstraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|
PairCorrelationConstraints contains classes for all constraints related to experimental pair correlation functions.

fullrmc.Constraints.PairCorrelationConstraints.PairCorrelationConstraint(experimentalData, dataWeights=None, weighting='atomicNumber', atomsWeight=None, scaleFactor=1.0, adjustScaleFactor=(0, 0.8, 1.2), shapeFuncParams=None, windowFunction=None, limits=None)¶Bases: fullrmc.Constraints.PairDistributionConstraints.PairDistributionConstraint
Controls the total pair correlation function (pcf) of the system noted as g(r). pcf indicates the probability of finding atomic pairs separated by the real space distance r. Theoretically g(r) oscillates about 1. Also \(g(r) \rightarrow 1\) when \(r \rightarrow \infty\) and it takes the exact value of zero for \(r\) shorter than the distance of the closest possible approach of pairs of atoms.
Pair correlation function g(r) and pair distribution function G(r) are directly related as in the following: \(g(r)=1+(\frac{G(r)}{4 \pi \rho_{0} r})\).
g(r) is calculated after binning all pair atomic distances into a weighted histograms of values \(n(r)\) from which local number densities are computed as in the following:
Where:
\(r\) is the distance between two atoms.
\(\rho_{i,j}(r)\) is the pair density function of atoms i and j.
\(\rho_{0}\) is the average number density of the system.
\(w_{i,j}\) is the relative weighting of atom types i and j.
\(N\) is the total number of atoms.
\(V\) is the volume of the system.
\(n_{i,j}(r)\) is the number of atoms i neighbouring j at a distance r.
\(v(r)\) is the annulus volume at distance r and of thickness dr.
\(N_{i,j}\) is the total number of atoms i and j in the system.
| Parameters: | Refer to PairDistributionConstraint |
|---|
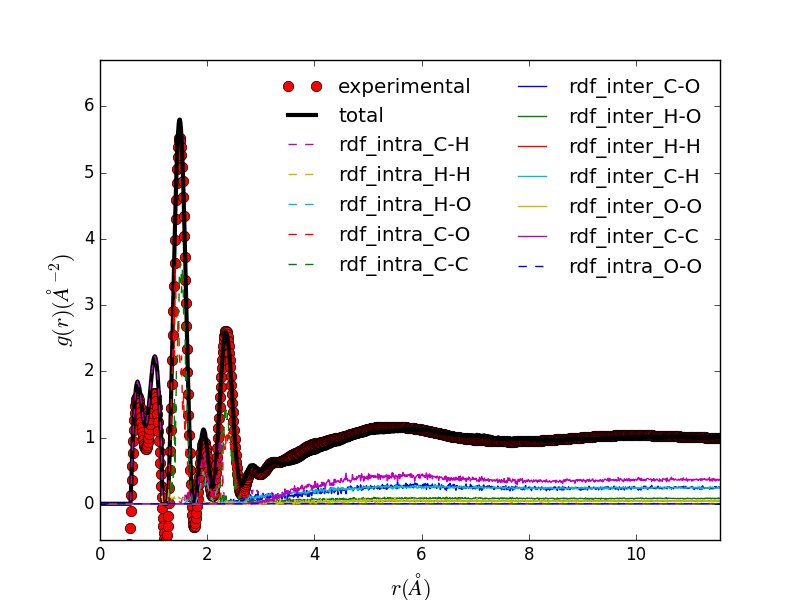
|
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Constraints.PairCorrelationConstraints import PairCorrelationConstraint
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# create and add constraint
PCC = PairCorrelationConstraint(experimentalData="pcf.dat", weighting="atomicNumber")
ENGINE.add_constraints(PCC)
update_standard_error()¶Compute and set constraint’s standardError.
get_adjusted_scale_factor(experimentalData, modelData, dataWeights, rho0)¶Overload to bring back g(r) to G(r) prior to fitting scale factor. g(r) -> 1 at high r and this will create a wrong scale factor. Overloading can be avoided but it’s better to for performance reasons
get_constraint_value(applyMultiframePrior=True)¶Get constraint’s data dictionary value.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_data(*args, **kwargs)¶Compute constraint’s data.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_before_move(realIndexes, relativeIndexes)¶Compute constraint’s data before move is executed.
| Parameters: |
|
|---|
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Compute constraint’s data after move is executed.
| Parameters: |
|
|---|
compute_as_if_amputated(realIndex, relativeIndex)¶Compute and return constraint’s data and standard error as if given atom is amputated.
| Parameters: |
|
|---|
plot(xlabelParams={'size': 10, 'xlabel': '$r(\\AA)$'}, ylabelParams={'size': 10, 'ylabel': '$g(r)(\\AA^{-2})$'}, **kwargs)¶Alias to ExperimentalConstraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|
StructureFactorConstraints contains classes for all constraints related experimental static structure factor functions.

fullrmc.Constraints.StructureFactorConstraints.StructureFactorConstraint(experimentalData, dataWeights=None, weighting='atomicNumber', atomsWeight=None, rmin=None, rmax=None, dr=None, scaleFactor=1.0, adjustScaleFactor=(0, 0.8, 1.2), windowFunction=None, limits=None)¶Bases: fullrmc.Core.Constraint.ExperimentalConstraint
Controls the Structure Factor noted as S(Q) and also called total-scattering structure function or Static Structure Factor. S(Q) is a dimensionless quantity and normalized such as the average value \(<S(Q)>=1\).
It is worth mentioning that S(Q) is nothing other than the normalized and corrected diffraction pattern if all experimental artefacts powder.
The computation of S(Q) is done through an inverse Sine Fourier transform of the computed pair distribution function G(r).
From an atomistic model and histogram point of view, G(r) is computed as the following:
g(r) is calculated after binning all pair atomic distances into a weighted histograms as the following:
Where:
\(Q\) is the momentum transfer.
\(r\) is the distance between two atoms.
\(\rho_{i,j}(r)\) is the pair density function of atoms i and j.
\(\rho_{0}\) is the average number density of the system.
\(w_{i,j}\) is the relative weighting of atom types i and j.
\(R(r)\) is the radial distribution function (rdf).
\(N\) is the total number of atoms.
\(V\) is the volume of the system.
\(n_{i,j}(r)\) is the number of atoms i neighbouring j at a distance r.
\(v(r)\) is the annulus volume at distance r and of thickness dr.
\(N_{i,j}\) is the total number of atoms i and j in the system.
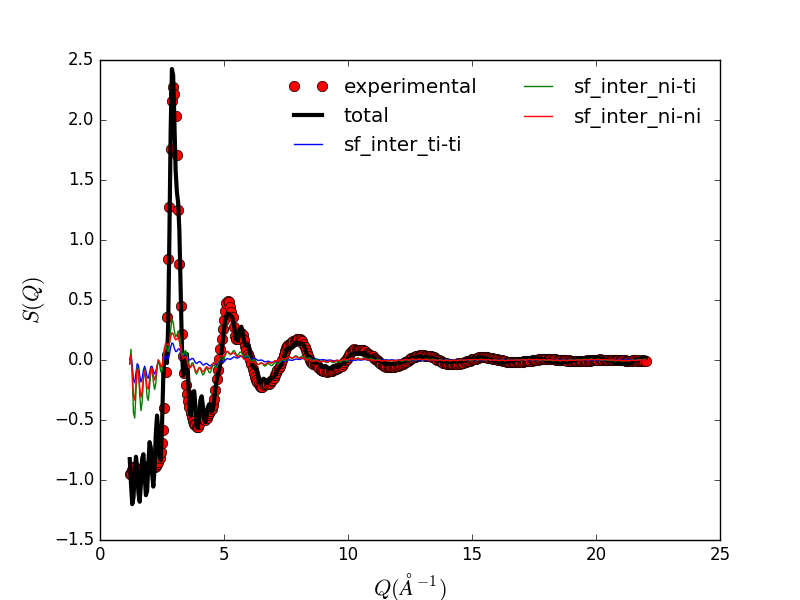
Reduced structure factor of memory shape Nickel-Titanium alloy. |
| Parameters: |
|
|---|
NB: If adjustScaleFactor first item (frequency) is 0, the scale factor will remain untouched and the limits minimum and maximum won’t be checked.
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Constraints.StructureFactorConstraints import StructureFactorConstraint
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# create and add constraint
SFC = StructureFactorConstraint(experimentalData="sq.dat", weighting="atomicNumber")
ENGINE.add_constraints(SFC)
rmin¶Histogram minimum distance.
rmax¶Histogram maximum distance.
dr¶Histogram bin size.
bin¶Computed histogram distance bin size.
minimumDistance¶Computed histogram minimum distance.
maximumDistance¶Computed histogram maximum distance.
qmin¶Experimental data reciprocal distances minimum.
qmax¶Experimental data reciprocal distances maximum.
dq¶Experimental data reciprocal distances bin size.
experimentalQValues¶Experimental data used q values.
histogramSize¶Histogram size
shellCenters¶Shells center array
shellVolumes¶Shells volume array
experimentalSF¶Experimental Structure Factor or S(q)
elementsPairs¶Elements pairs
atomsWeight¶Custom atoms weight
weighting¶Elements weighting definition.
weightingScheme¶Elements weighting scheme.
windowFunction¶Convolution window function.
Gr2SqMatrix¶G(r) to S(q) transformation matrix.
listen(message, argument=None)¶Listens to any message sent from the Broadcaster.
| Parameters: |
|
|---|
set_rmin(rmin)¶Set rmin value.
| Parameters: |
|
|---|
set_rmax(rmax)¶Set rmax value.
| Parameters: |
|
|---|
set_dr(dr)¶Set dr value.
| Parameters: |
|
|---|
set_weighting(weighting)¶Set elements weighting. It must be a valid entry of pdbparser atom’s database.
| Parameters: |
|
|---|
set_atoms_weight(atomsWeight)¶Custom set atoms weight. This is the way to setting a atoms weights different than the given weighting scheme.
| Parameters: |
|
|---|
set_window_function(windowFunction)¶Set convolution window function.
| Parameters: |
|
|---|
set_experimental_data(experimentalData)¶Set constraint’s experimental data.
| Parameters: |
|
|---|
set_limits(limits)¶Set the reciprocal distance limits (qmin, qmax).
| Parameters: |
|
|---|
update_standard_error()¶Compute and set constraint’s standardError.
check_experimental_data(experimentalData)¶Check whether experimental data is correct.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_standard_error(modelData)¶Compute the standard error (StdErr) as the squared deviations between model computed data and the experimental ones.
Where:
\(N\) is the total number of experimental data points.
\(W_{i}\) is the data point weight. It becomes equivalent to 1 when dataWeights is set to None.
\(Y(X_{i})\) is the experimental data point \(X_{i}\).
\(F(X_{i})\) is the computed from the model data \(X_{i}\).
| Parameters: |
|
|---|---|
| Returns: |
|
get_adjusted_scale_factor(experimentalData, modelData, dataWeights)¶Overload to reduce S(q) prior to fitting scale factor. S(q) -> 1 at high q and this will create a wrong scale factor. Overloading can be avoided but it’s better to for performance reasons
get_constraint_value(applyMultiframePrior=True)¶Compute all partial Structure Factor (SQs).
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraint_original_value()¶Compute all partial Pair Distribution Functions (PDFs).
| Returns: |
|
|---|
compute_data(*args, **kwargs)¶Compute constraint’s data.
| Parameters: |
|
|---|---|
| Returns: |
|
compute_before_move(realIndexes, relativeIndexes)¶Compute constraint before move is executed
| Parameters: |
|
|---|
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Compute constraint after move is executed
| Parameters: |
|
|---|
accept_move(realIndexes, relativeIndexes)¶Accept move
| Parameters: |
|
|---|
reject_move(realIndexes, relativeIndexes)¶Reject move
| Parameters: |
|
|---|
compute_as_if_amputated(realIndex, relativeIndex)¶Compute and return constraint’s data and standard error as if given atom is amputated.
| Parameters: |
|
|---|
accept_amputation(realIndex, relativeIndex)¶Accept amputated atom and sets constraints data and standard error accordingly.
| Parameters: |
|
|---|
reject_amputation(realIndex, relativeIndex)¶Reject amputated atom and set constraint’s data and standard error accordingly.
| Parameters: |
|
|---|
plot(xlabelParams={'size': 10, 'xlabel': '$Q(\\AA^{-1})$'}, ylabelParams={'size': 10, 'ylabel': '$S(Q)$'}, **kwargs)¶Alias to ExperimentalConstraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|
fullrmc.Constraints.StructureFactorConstraints.ReducedStructureFactorConstraint(experimentalData, dataWeights=None, weighting='atomicNumber', atomsWeight=None, rmin=None, rmax=None, dr=None, scaleFactor=1.0, adjustScaleFactor=(0, 0.8, 1.2), windowFunction=None, limits=None)¶Bases: fullrmc.Constraints.StructureFactorConstraints.StructureFactorConstraint
The Reduced Structure Factor that we will also note S(Q) is exactly the same quantity as the Structure Factor but with the slight difference that it is normalized to 0 rather than 1 and therefore \(<S(Q)>=0\).
The computation of S(Q) is done through a Sine inverse Fourier transform of the computed pair distribution function noted as G(r).
The only reason why the Reduced Structure Factor is implemented, is because many experimental data are treated in this form. And it is just convenient not to manipulate the experimental data every time.
get_adjusted_scale_factor(experimentalData, modelData, dataWeights)¶dummy overload that does exactly the same thing
plot(xlabelParams={'size': 10, 'xlabel': '$Q(\\AA^{-1})$'}, ylabelParams={'size': 10, 'ylabel': '$S(Q)-1$'}, **kwargs)¶Alias to ExperimentalConstraint.plot with additional parameters
| Additional/Adjusted Parameters: | |
|---|---|
|
|