It contains a collection of methods and classes that are useful for the package.
fullrmc.Core.Collection.get_caller_frames(engine, frame, subframeToAll, caller)¶Get list of frames for a function caller.
| Parameters: |
|
|---|---|
| Returns: |
|
import inspect
from fullrmc.Core.Collection import get_caller_frames
# Assuming self is a constraint and get_caller_frames is called from within a method ...
usedIncluded, frame, allFrames = get_caller_frames(engine=self.engine,
frame='frame_name',
subframeToAll=True,
caller="%s.%s"%(self.__class__.__name__,inspect.stack()[0][3]) )
fullrmc.Core.Collection.get_real_elements_weight(elements, weightsDict, weighting)¶Get elements weights given a dictionary of weights and a weighting scheme. If element weight is not defined in weightsDict then weight is fetched from pdbparser elements database using weighting scheme.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.raise_if_collected(func)¶Constraints method decorator that raises an error whenever the method is called and the system has atoms that were removed.
fullrmc.Core.Collection.reset_if_collected_out_of_date(func)¶Constraints method decorator that resets the constraint whenever the method is called and the system has atoms that were removed.
fullrmc.Core.Collection.is_number(number)¶Check if number is convertible to float.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.is_integer(number, precision=1e-09)¶Check if number is convertible to integer.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.get_elapsed_time(start, format='%d days, %d hours, %d minutes, %d seconds')¶Get formatted time elapsed.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.get_memory_usage()¶Get current process memory usage. This is method requires psutils to be installed.
| Returns: |
|
|---|
fullrmc.Core.Collection.get_path(key=None)¶Get all paths information needed about the running script and python executable path.
| Parameters: | #. key (None, string): the path to return. If not None is given, it can take any of the following:
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.rebin(data, bin=0.05, check=False)¶Re-bin 2D data of shape (N,2). In general, fullrmc requires equivalently spaced experimental data bins. This function can be used to recompute any type of experimental data according to a set bin size.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.smooth(data, winLen=11, window='hanning', check=False)¶Smooth 1D data using window function and length.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.get_random_perpendicular_vector(vector)¶Get random normalized perpendicular vector to a given vector.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.get_principal_axis(coordinates, weights=None)¶Calculate principal axis of a set of atoms coordinates.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.get_rotation_matrix(rotationVector, angle)¶Calculate the rotation (3X3) matrix about an axis (rotationVector) by a rotation angle.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.rotate(xyzArray, rotationMatrix)¶Rotate (N,3) numpy.array using a rotation matrix. The array itself will be rotated and not a copy of it.
| Parameters: |
|
|---|
fullrmc.Core.Collection.get_orientation_matrix(arrayAxis, alignToAxis)¶Get the rotation matrix that aligns arrayAxis to alignToAxis
| Parameters: |
|
|---|
fullrmc.Core.Collection.orient(xyzArray, arrayAxis, alignToAxis)¶Rotates xyzArray using the rotation matrix that rotates and aligns arrayAxis to alignToAXis.
| Parameters: |
|
|---|
fullrmc.Core.Collection.get_superposition_transformation(refArray, array, check=False)¶Calculate the rotation tensor and the translations that minimizes the root mean square deviation between an array of vectors and a reference array.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.superpose_array(refArray, array, check=False)¶Superpose arrays by calculating the rotation matrix and the translations that minimize the root mean square deviation between and array of vectors and a reference array.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.generate_random_vector(minAmp, maxAmp)¶Generate random vector in 3D.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.generate_points_on_sphere(thetaFrom, thetaTo, phiFrom, phiTo, npoints=1, check=False)¶Generate random points on a sphere of radius 1. Points are generated using spherical coordinates arguments as in figure below. Theta [0,Pi] is the angle between the generated point and Z axis. Phi [0,2Pi] is the angle between the generated point and x axis.
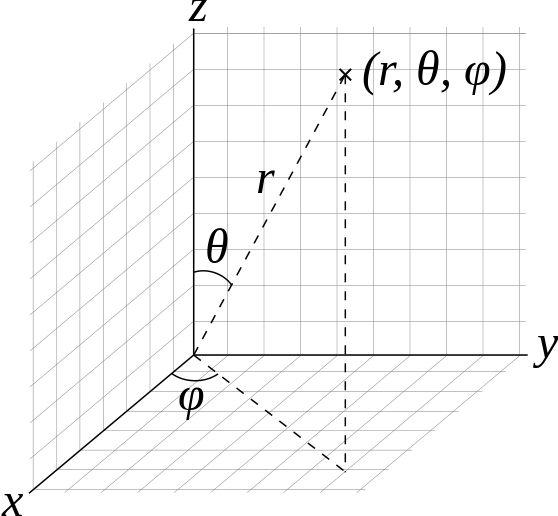
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.find_extrema(x, max=True, min=True, strict=False, withend=False)¶Get a vector extrema indexes and values.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.convert_Gr_to_gr(Gr, minIndex)¶Converts G(r) to g(r) by computing the following \(g(r)=1+(\frac{G(r)}{4 \pi \rho_{0} r})\)
| Parameters: |
|
|---|---|
| Returns: |
|
To visualize convertion
# peak indexes can be different, adjust according to your data
minPeaksIndex = [1,3,4]
minimas, slope, rho0, gr = convert_Gr_to_gr(Gr, minIndex=minPeaksIndex)
print('slope: %s --> rho0: %s'%(slope,rho0))
import matplotlib.pyplot as plt
line = np.transpose( [[0, Gr[-1,0]], [0, slope*Gr[-1,0]]] )
plt.plot(Gr[:,0],Gr[:,1], label='G(r)')
plt.plot(minimas[:,0], minimas[:,1], 'o', label='minimas')
plt.plot(line[:,0], line[:,1], label='density')
plt.plot(gr[:,0],gr[:,1], label='g(r)')
plt.legend()
plt.show()
fullrmc.Core.Collection.generate_vectors_in_solid_angle(direction, maxAngle, numberOfVectors=1, check=False)¶Generate random vectors that satisfy angle condition with a direction vector. Angle between any generated vector and direction must be smaller than given maxAngle.
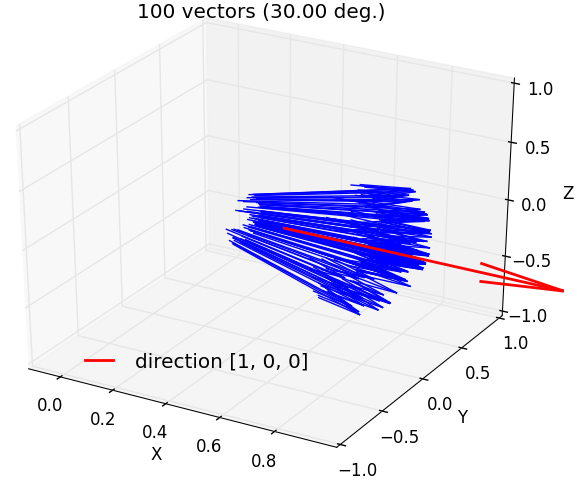
a) 100 vectors generated around OX axis within a maximum angle separation of 30 degrees. |
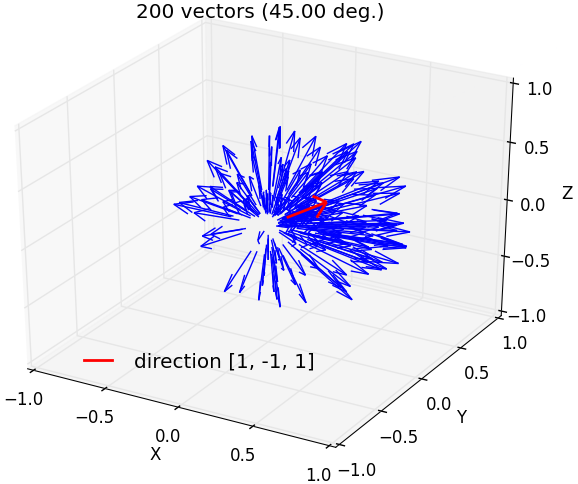
b) 200 vectors generated around [1,-1,1] axis within a maximum angle separation of 45 degrees. |
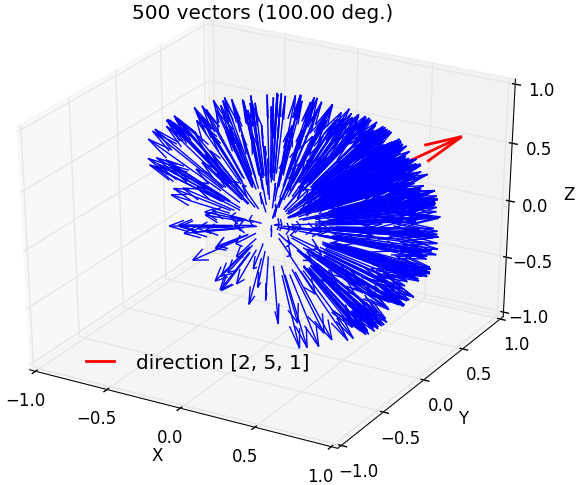
b) 500 vectors generated around [2,5,1] axis within a maximum angle separation of 100 degrees. |
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Collection.gaussian(x, center=0, FWHM=1, normalize=True, check=True)¶Compute the normal distribution or gaussian distribution of a given vector. The probability density of the gaussian distribution is: \(f(x,\mu,\sigma) = \frac{1}{\sigma\sqrt{2\pi}} e^{\frac{-(x-\mu)^{2}}{2\sigma^2}}\)
Where:
| Parameters: |
|
|---|
fullrmc.Core.Collection.step_function(x, center=0, FWHM=0.1, height=1, check=True)¶Compute a step function as the cumulative summation of a gaussian distribution of a given vector.
| Parameters: |
|
|---|
fullrmc.Core.Collection.ListenerBase¶Bases: object
All listeners base class.
listenerId¶Listener unique id set at initialization
listen(message, argument=None)¶Listens to any message sent from the Broadcaster.
| Parameters: |
|
|---|
fullrmc.Core.Collection.Broadcaster¶Bases: object
A broadcaster broadcasts a message to all registered listener.
listeners¶Listeners list copy.
add_listener(listener)¶Add listener to the list of listeners.
| Parameters: |
|
|---|
remove_listener(listener)¶Remove listener to the list of listeners.
| Parameters: |
|
|---|
broadcast(message, arguments=None)¶Broadcast a message to all the listeners
| Parameters: |
|
|---|
fullrmc.Core.Collection.RandomFloatGenerator(lowerLimit, upperLimit)¶Bases: object
Generate random float number between a lower and an upper limit.
| Parameters: |
|
|---|
lowerLimit¶Lower limit of the number generation.
upperLimit¶Upper limit of the number generation.
rang¶Range defined as upperLimit-lowerLimit.
set_lower_limit(lowerLimit)¶Set lower limit.
| Parameters: |
|
|---|
set_upper_limit(upperLimit)¶Set upper limit.
| Parameters: |
|
|---|
generate()¶Generate a random float number between lowerLimit and upperLimit.
fullrmc.Core.Collection.BiasedRandomFloatGenerator(lowerLimit, upperLimit, weights=None, biasRange=None, biasFWHM=None, biasHeight=1, unbiasRange=None, unbiasFWHM=None, unbiasHeight=None, unbiasThreshold=1)¶Bases: fullrmc.Core.Collection.RandomFloatGenerator
Generate biased random float number between a lower and an upper limit. To bias the generator at a certain number, a bias gaussian is added to the weights scheme at the position of this particular number.
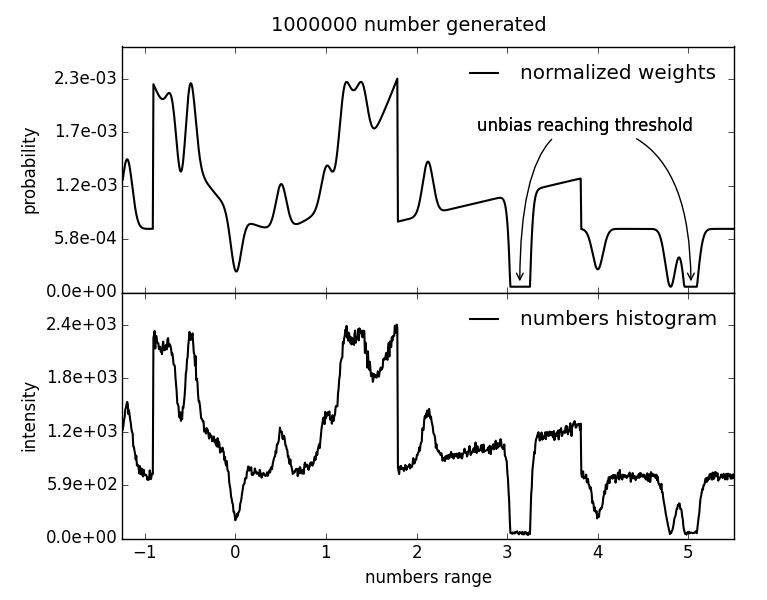
| Parameters: |
|
|---|
originalWeights¶Original weights as initialized.
weights¶Current value weights vector.
scheme¶Numbers generation scheme.
bins¶Number of bins that is equal to the length of weights vector.
binWidth¶Bin width defining the resolution of the biased random number generation.
bias¶Bias step-function.
biasGuassian¶Bias gaussian function.
biasRange¶Bias gaussian extent range.
biasBins¶Bias gaussian number of bins.
biasFWHM¶Bias gaussian Full Width at Half Maximum.
biasFWHMBins¶Bias gaussian Full Width at Half Maximum number of bins.
unbias¶Unbias step-function.
unbiasGuassian¶Unbias gaussian function.
unbiasRange¶Unbias gaussian extent range.
unbiasBins¶Unbias gaussian number of bins.
unbiasFWHM¶Unbias gaussian Full Width at Half Maximum.
unbiasFWHMBins¶Unbias gaussian Full Width at Half Maximum number of bins.
set_weights(weights=None)¶Set generator’s weights.
| Parameters: |
|
|---|
set_bias(biasRange, biasFWHM, biasHeight)¶Set generator’s bias gaussian function
| Parameters: |
|
|---|
set_unbias(unbiasRange, unbiasFWHM, unbiasHeight, unbiasThreshold)¶Set generator’s unbias gaussian function
| Parameters: |
|
|---|
bias_scheme_by_index(index, scaleFactor=None, check=True)¶Bias the generator’s scheme using the defined bias gaussian function at the given index.
| Parameters: |
|
|---|
bias_scheme_at_position(position, scaleFactor=None, check=True)¶Bias the generator’s scheme using the defined bias gaussian function at the given number.
| Parameters: |
|
|---|
unbias_scheme_by_index(index, scaleFactor=None, check=True)¶Unbias the generator’s scheme using the defined bias gaussian function at the given index.
| Parameters: |
|
|---|
unbias_scheme_at_position(position, scaleFactor=None, check=True)¶Unbias the generator’s scheme using the defined bias gaussian function at the given number.
| Parameters: |
|
|---|
generate()¶Generate a random float number between the biased range lowerLimit and upperLimit.
fullrmc.Core.Collection.RandomIntegerGenerator(lowerLimit, upperLimit)¶Bases: object
Generate random integer number between a lower and an upper limit.
| Parameters: |
|
|---|
lowerLimit¶Lower limit of the number generation.
upperLimit¶Upper limit of the number generation.
rang¶The range defined as upperLimit-lowerLimit
set_lower_limit(lowerLimit)¶Set lower limit.
| Parameters: |
|
|---|
set_upper_limit(upperLimit)¶Set upper limit.
| Parameters: |
|
|---|
generate()¶Generate a random integer number between lowerLimit and upperLimit.
fullrmc.Core.Collection.BiasedRandomIntegerGenerator(lowerLimit, upperLimit, weights=None, biasHeight=1, unbiasHeight=None, unbiasThreshold=1)¶Bases: fullrmc.Core.Collection.RandomIntegerGenerator
Generate biased random integer number between a lower and an upper limit. To bias the generator at a certain number, a bias height is added to the weights scheme at the position of this particular number.
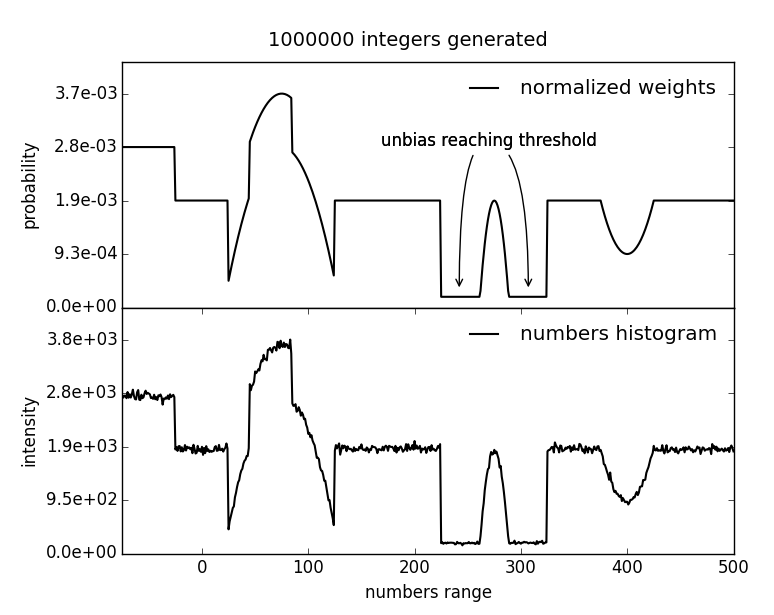
| Parameters: |
|
|---|
originalWeights¶Original weights as initialized.
weights¶Current value weights vector.
scheme¶Numbers generation scheme.
bins¶Number of bins that is equal to the length of weights vector.
set_weights(weights)¶Set the generator integer numbers weights.
| Parameters: |
|
|---|
set_bias_height(biasHeight)¶Set weight bias intensity.
| Parameters: |
|
|---|
set_unbias_height(unbiasHeight)¶Set weight unbias intensity.
| Parameters: |
|
|---|
set_unbias_threshold(unbiasThreshold)¶Set weight unbias threshold.
| Parameters: |
|
|---|
bias_scheme_by_index(index, scaleFactor=None, check=True)¶Bias the generator’s scheme at the given index.
| Parameters: |
|
|---|
bias_scheme_at_position(position, scaleFactor=None, check=True)¶Bias the generator’s scheme at the given number.
| Parameters: |
|
|---|
unbias_scheme_by_index(index, scaleFactor=None, check=True)¶Unbias the generator’s scheme at the given index.
| Parameters: |
|
|---|
unbias_scheme_at_position(position, scaleFactor=None, check=True)¶Unbias the generator’s scheme using the defined bias gaussian function at the given number.
| Parameters: |
|
|---|
generate()¶Generate a random intger number between the biased range lowerLimit and upperLimit.
fullrmc.Core.Collection.generate_random_float()¶random() -> x in the interval [0, 1).
Constraint contains parent classes for all constraints. A Constraint is used to set certain rules for the stochastic engine to evolve the atomic system. Therefore it has become possible to fully customize and set any possibly imaginable rule.
fullrmc.Core.Constraint.Constraint¶Bases: fullrmc.Core.Collection.ListenerBase
A constraint is used to direct the evolution of the atomic configuration towards the desired and most meaningful one.
constraintId¶Constraint unique ID create at instanciation time.
constraintName¶Constraints unique name in engine given when added to engine.
engine¶Stochastic fullrmc’s engine instance.
usedFrame¶Get used frame in engine. If None then engine is not defined yet
computationCost¶Computation cost number.
state¶Constraint’s state.
tried¶Constraint’s number of tried moves.
accepted¶Constraint’s number of accepted moves.
used¶Constraint’s used flag. Defines whether constraint is used in the stochastic engine at runtime or set inactive.
varianceSquared¶Constraint’s varianceSquared used in the stochastic engine at runtime to calculate the total constraint’s standard error.
standardError¶Constraint’s standard error value.
originalData¶Constraint’s original data calculated upon initialization.
data¶Constraint’s current calculated data.
activeAtomsDataBeforeMove¶Constraint’s current calculated data before last move.
activeAtomsDataAfterMove¶Constraint’s current calculated data after last move.
afterMoveStandardError¶Constraint’s current calculated StandardError after last move.
amputationData¶Constraint’s current calculated data after amputation.
amputationStandardError¶Constraint’s current calculated StandardError after amputation.
multiframeWeight¶Get constraint weight towards total in a multiframe system.
is_in_engine(engine)¶Get whether constraint is already in defined and added to engine. It can be the same exact instance or a repository pulled instance of the same constraintId
| Parameters: |
|
|---|---|
| Returns: |
|
set_variance_squared(value, frame=None)¶Set constraint’s variance squared that is used in the computation of the total stochastic engine standard error.
| Parameters: |
|
|---|
set_computation_cost(value, frame=None)¶Set constraint’s computation cost value. This is used at stochastic engine runtime to minimize computations and enhance performance by computing less costly constraints first. At every step, constraints will be computed in order starting from the less to the most computationally costly. Therefore upon rejection of a step because of an unsatisfactory rigid constraint, the left un-computed constraints at this step are guaranteed to be the most time coslty ones.
| Parameters: |
|
|---|
set_used(*args, **kwargs)¶Set used flag.
| Parameters: |
|
|---|
set_state(value)¶Set constraint’s state. When constraint’s state and stochastic engine’s state don’t match, constraint’s data must be re-calculated.
| Parameters: |
|
|---|
set_tried(value)¶Set constraint’s number of tried moves.
| Parameters: |
|
|---|
increment_tried()¶Increment number of tried moves.
set_accepted(value)¶Set constraint’s number of accepted moves.
| Parameters: |
|
|---|
increment_accepted()¶Increment constraint’s number of accepted moves.
set_standard_error(value)¶Set constraint’s standardError value.
| Parameters: |
|
|---|
set_data(value)¶Set constraint’s data value.
| Parameters: |
|
|---|
set_active_atoms_data_before_move(value)¶Set constraint’s before move happens active atoms data value.
| Parameters: |
|
|---|
set_active_atoms_data_after_move(value)¶Set constraint’s after move happens active atoms data value.
| Parameters: |
|
|---|
set_after_move_standard_error(value)¶Set constraint’s standard error value after move happens.
| Parameters: |
|
|---|
set_amputation_data(value)¶Set constraint’s after amputation data.
| Parameters: |
|
|---|
set_amputation_standard_error(value)¶Set constraint’s standardError after amputation.
| Parameters: |
|
|---|
reset_constraint(reinitialize=True, flags=False, data=False, frame=None)¶Reset constraint.
| Parameters: |
|
|---|
update_standard_error()¶Compute and set constraint’s standard error by calling compute_standard_error method and passing constraint’s data.
get_constraints_properties(frame)¶Get a dictionary look up table of constraint’s properties
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraint_value()¶Method must be overloaded in children classes.
get_constraint_original_value()¶Method must be overloaded in children classes.
compute_standard_error()¶Method must be overloaded in children classes.
compute_data(update=True)¶Method must be overloaded in children classes.
compute_before_move(realIndexes, relativeIndexes)¶Method must be overloaded in children classes.
compute_after_move(realIndexes, relativeIndexes, movedBoxCoordinates)¶Method must be overloaded in children classes.
accept_move(realIndexes, relativeIndexes)¶Method must be overloaded in children classes.
reject_move(realIndexes, relativeIndexes)¶Method must be overloaded in children classes.
compute_as_if_amputated(realIndex, relativeIndex)¶Method must be overloaded in children classes.
compute_as_if_inserted(realIndex, relativeIndex)¶Method must be overloaded in children classes.
accept_amputation(realIndex, relativeIndex)¶Method must be overloaded in children classes.
reject_amputation(realIndex, relativeIndex)¶Method must be overloaded in children classes.
accept_insertion(realIndex, relativeIndex)¶Method must be overloaded in children classes.
reject_insertion(realIndex, relativeIndex)¶Method must be overloaded in children classes.
export(fileName, frame=None, format='%s', delimiter='\t', comments='#')¶Export constraint data to text file or to an archive of files.
| Parameters: |
|
|---|
plot(frame=None, axes=None, subAdParams={'bottom': None, 'hspace': 0.4, 'left': None, 'right': None, 'top': None, 'wspace': None}, dataParams={'label': 'Y', 'linewidth': 2}, xlabelParams={'size': 10, 'xlabel': 'Core-Shell atoms'}, ylabelParams={'size': 10, 'ylabel': 'Coordination number'}, xticksParams={'fontsize': 8, 'rotation': 90}, yticksParams={'fontsize': 8, 'rotation': 0}, legendParams={'fontsize': 8, 'frameon': False, 'loc': 'upper right', 'ncol': 1}, titleParams={'fontsize': 10, 'label': '@{frame} (${numberOfRemovedAtoms:.1f}$ $rem.$ $at.$) $Std.Err.={standardError:.3f}$ - $used$ $({used})$'}, gridParams=None, show=True, **paramsKwargs)¶Plot constraint data. This can be overloaded in children classes.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.Constraint.ExperimentalConstraint(experimentalData, dataWeights=None, scaleFactor=1.0, adjustScaleFactor=(0, 0.8, 1.2))¶Bases: fullrmc.Core.Constraint.Constraint
Experimental constraint is any constraint related to experimental data.
| Parameters: |
|
|---|
NB: If adjustScaleFactor first item (frequency) is 0, the scale factor will remain untouched and the limits minimum and maximum won’t be checked.
experimentalData¶Experimental data of the constraint.
dataWeights¶Experimental data points weight
multiframeWeight¶Get constraint weight towards total in a multiframe system.
multiframePrior¶Get constraint multiframe prior array.
scaleFactor¶Constraint’s scaleFactor.
adjustScaleFactor¶Adjust scale factor tuple.
adjustScaleFactorFrequency¶Scale factor adjustment frequency.
adjustScaleFactorMinimum¶Scale factor adjustment minimum number allowed.
adjustScaleFactorMaximum¶Scale factor adjustment maximum number allowed.
limits¶Used data X limits.
limitsIndexStart¶Used data start index as calculated from limits.
limitsIndexEnd¶Used data end index as calculated from limits.
set_scale_factor(scaleFactor)¶Set the scale factor. This method doesn’t allow specifying frames. It will target used frame only.
| Parameters: |
|
|---|
set_adjust_scale_factor(adjustScaleFactor)¶Set adjust scale factor. This method doesn’t allow specifying frames. It will target used frame only.
| Parameters: |
|
|---|
set_experimental_data(experimentalData)¶Set the constraint’s experimental data. This method will raise an error if called after adding constraint to stochastic engine.
| Parameters: |
|
|---|
set_data_weights(dataWeights, frame=None)¶Set experimental data points weight. Data weights will be automatically normalized.
| Parameters: |
|
|---|
check_experimental_data(experimentalData)¶Checks the constraint’s experimental data This method must be overloaded in all experimental constraint sub-classes.
| Parameters: |
|
|---|
fit_scale_factor(experimentalData, modelData, dataWeights)¶The best scale factor value is computed by minimizing \(E=sM\).
This method doesn’t allow specifying frames. It will target used frame only.
| Parameters: |
|
|---|---|
| Returns: |
|
NB: This method won’t update the internal scale factor value of the constraint. It always computes the best scale factor given experimental and atomic model data.
get_adjusted_scale_factor(experimentalData, modelData, dataWeights)¶Checks if scale factor should be updated according to the given scale factor frequency and engine’s accepted steps. If adjustment is due, a new scale factor will be computed using fit_scale_factor method, otherwise the the constraint’s scale factor will be returned.
| Parameters: |
|
|---|---|
| Returns: | #. scaleFactor (number): Constraint’s scale factor or the new scale factor fit value. |
NB: This method WILL NOT UPDATE the internal scale factor value of the constraint.
compute_standard_error(experimentalData, modelData)¶Compute the squared deviation between modal computed data and the experimental ones.
Where:
\(N\) is the total number of experimental data points.
\(W_{i}\) is the data point weight. It becomes equivalent to 1 when dataWeights is set to None.
\(Y(X_{i})\) is the experimental data point \(X_{i}\).
\(F(X_{i})\) is the computed from the model data \(X_{i}\).
| Parameters: |
|
|---|---|
| Returns: |
|
get_constraints_properties(frame)¶Get a dictionary look up table of constraint’s properties that are needed to plot or export
| Parameters: |
|
|---|---|
| Returns: |
|
plot(frame=None, axes=None, asMesoscopic=False, intra=True, inter=True, shapeFunc=True, subAdParams={'bottom': None, 'hspace': 0.4, 'left': None, 'right': None, 'top': None, 'wspace': None}, totParams={'color': 'black', 'label': 'total', 'linewidth': 3.0, 'zorder': 1}, expParams={'color': 'red', 'label': 'experimental', 'marker': 'o', 'markersize': 7.5, 'markevery': 1, 'zorder': 0}, noWParams={'color': 'black', 'label': 'total - no window', 'linewidth': 1.0, 'zorder': 1}, shaParams={'color': 'black', 'label': 'shape function', 'linestyle': 'dashed', 'linewidth': 1.0, 'zorder': 2}, parParams={'linewidth': 1.0, 'markersize': 5, 'markevery': 5, 'zorder': 3}, xlabelParams={'size': 10, 'xlabel': 'X'}, ylabelParams={'size': 10, 'ylabel': 'Y'}, xticksParams={'fontsize': 8, 'rotation': 0}, yticksParams={'fontsize': 8, 'rotation': 0}, legendParams={'fontsize': 8, 'frameon': False, 'loc': 'upper right', 'ncol': 2}, titleParams={'fontsize': 10, 'label': '@{frame} (${numberOfRemovedAtoms:.1f}$ $rem.$ $at.$) $Std.Err.={standardError:.3f}$\n$scale$ $factor$=${scaleFactor:.2f}$ - $multiframe$ $weight$=${multiframeWeight:.3f}$ - $used$ $({used})$'}, gridParams=None, show=True, **paramsKwargs)¶Plot constraint data. This can be overloaded in children classes.
| Parameters: |
|
|---|---|
| Returns: |
|
plot_multiframe_weights(frame, ax=None, titleFormat='@{frame} [subframes probability distribution]', show=True)¶plot multiframe subframes weight distribution histogram
| Parameters: |
|
|---|---|
| Returns: |
|
export(fileName, frame=None, format='%12.5f', delimiter='\t', comments='#')¶Export constraint data to text file or to an archive of files.
| Parameters: |
|
|---|
fullrmc.Core.Constraint.SingularConstraint¶Bases: fullrmc.Core.Constraint.Constraint
A singular constraint is a constraint that doesn’t allow multiple instances in the same engine.
is_singular(engine)¶Get whether only one instance of this constraint type is present in the stochastic engine. True for only itself found, False for other instance of the same __class__.__name__ or constraintId.
| Parameters: |
|
|---|---|
| Returns: |
|
assert_singular(engine)¶Checks whether only one instance of this constraint type is present in the stochastic engine. Raises Exception if multiple instances are present.
fullrmc.Core.Constraint.RigidConstraint(rejectProbability)¶Bases: fullrmc.Core.Constraint.Constraint
A rigid constraint is a constraint that doesn’t count into the total standard error of the stochastic Engine. But it’s internal standard error must monotonously decrease or remain the same from one engine step to another. If standard error of an rigid constraint increases the step will be rejected even before engine’s new standardError get computed.
| Parameters: |
|
|---|
rejectProbability¶Rejection probability.
set_reject_probability(rejectProbability)¶Set the rejection probability. This method doesn’t allow specifying frames. It will target used frame only.
| Parameters: |
|
|---|
should_step_get_rejected(standardError)¶Given a standard error, return whether to keep or reject new standard error according to the constraint reject probability.
| Parameters: | #. standardError (number): The standard error to compare with the Constraint standard error |
|---|---|
| Return: |
|
should_step_get_accepted(standardError)¶Given a standard error, return whether to keep or reject new standard error according to the constraint reject probability.
| Parameters: |
|
|---|---|
| Return: |
|
fullrmc.Core.Constraint.randfloat()¶random() -> x in the interval [0, 1).
Group contains parent classes for all groups. A Group is a set of atoms indexes used to gather atoms and apply actions such as moves upon them. Therefore it has become possible to fully customize and separate atoms to groups and perform stochastic actions on groups rather than on single atoms.
fullrmc.Core.Group.Group(indexes, moveGenerator=None, refine=False, name='')¶Bases: object
A Group is a set of atoms indexes container.
| Parameters: |
|
|---|
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Core.Group import Group
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# Add constraints ...
# re-define groups as atoms.
groups = [Group([idx]) for idx in ENGINE.pdb.indexes]
ENGINE.set_groups( groups )
# Re-define groups generators as needed ... By default AtomsRemoveGenerator is used.
indexes¶Atoms index array.
moveGenerator¶Group’s move generator instance.
refine¶Refine flag.
name¶groud user defined name.
set_refine(refine)¶Set the selector refine flag.
| Parameters: |
|
|---|
set_name(name)¶Set the group’s name.
| Parameters: |
|
|---|
set_indexes(indexes)¶Set group atoms index. Indexes redundancy is not checked and indexes order is preserved.
| Parameters: |
|
|---|
set_move_generator(generator)¶Set group move generator.
| Parameters: |
|
|---|
fullrmc.Core.Group.EmptyGroup(*args, **kwargs)¶Bases: fullrmc.Core.Group.Group
Empty group is a special group that takes no atoms indexes. It’s mainly used to remove atoms from system upon fitting.
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Core.Group import EmptyGroup
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# Add constraints ...
# re-define groups and set a single empty group
ENGINE.set_groups( EmptyGroup() )
# Re-define groups generators as needed ... By default RemoveGenerator is used.
moveGenerator¶Group’s move generator instance.
indexes¶Always returns None for EmptyGroup
set_move_generator(generator)¶Set group move generator.
| Parameters: |
|
|---|
set_indexes(indexes)¶Sets the group indexes. For an EmptyGroup, this method will disregard given indexes argument and will always set indexes property to None.
| Parameters: |
|
|---|
GroupSelector contains parent classes for all group selectors. A GroupSelector is used at the stochastic engine’s runtime to select groups upon which a move will be applied. Therefore it has become possible to fully customize the selection of groups of atoms and to choose when and how frequently a group can be chosen to perform a move upon.
fullrmc.Core.GroupSelector.GroupSelector(engine=None)¶Bases: object
Group selector is the parent class that selects groups to perform moves at stochastic engine’s runtime.
| Parameters: |
|
|---|
engine¶Stochastic engine’s instance.
refine¶Get refine flag value. It will always return False because refine is a property of RecursiveGroupSelector instances only.
explore¶Get explore flag value. It will always return False because explore is a property of RecursiveGroupSelector instances only.
willSelect¶Get whether next step a new selection is occur or still the same group is going to be selected again. It will always return True because recurrence is a property of RecursiveGroupSelector instances only.
willRecur¶Get whether next step the same group will be returned. It will always return False because this is a property of RecursiveGroupSelector instances only.
willRefine¶Get whether selection is recurring and refine flag is True. It will always return False because recurrence is a property of RecursiveGroupSelector instances only.
willExplore¶Get whether selection is recurring and explore flag is True. It will always return False because recurrence is a property of RecursiveGroupSelector instances only.
isNewSelection¶Get whether the last step a new selection was made. It will always return True because recurrence is a property of RecursiveGroupSelector instances only.
isRecurring¶Get whether the last step the same group was returned. It will always return False because this is a property of RecursiveGroupSelector instances only.
isRefining¶Get whether selection is recurring and refine flag is True. It will always return False because recurrence is a property of RecursiveGroupSelector instances only.
isExploring¶Get whether selection is recurring and explore flag is True. It will always return False because recurrence is a property of RecursiveGroupSelector instances only.
set_engine(engine)¶Set selector’s stochastic engine instance.
| Parameters: |
|
|---|
select_index()¶This method must be overloaded in every GroupSelector sub-class
| Returns: |
|
|---|
move_accepted(index)¶This method is called by the stochastic engine when a move generated on a group is accepted. This method is empty must be overloaded when needed.
| Parameters: |
|
|---|
move_rejected(index)¶This method is called by the stochastic engine when a move generated on a group is rejected. This method is empty must be overloaded when needed.
| Parameters: |
|
|---|
fullrmc.Core.GroupSelector.RecursiveGroupSelector(selector, recur=10, override=True, refine=False, explore=False)¶Bases: fullrmc.Core.GroupSelector.GroupSelector
Recursive selector is the only selector that can use the recursive property on a selection. It is used as a wrapper around a GroupSelector instance.
| Parameters: |
|
|---|
NB: refine and explore flags can’t both be set to True at the same time. When this happens refine flag gets automatically switched to False. The usage of those flags is very important because they allow groups of atoms to go out of local minima in the energy surface. The way traditional reverse mote carlo works is by minimizing the total energy of the system (error) using gradient descent method. Using of those flags allows the system to go up hill in the energy surface searching for other lower minimas, while always conserving the lowest energy state found and not changing the system structure until a better structure with smaller error is found.
The following video compares the Reverse Monte Carlo traditional fitting mode with fullrmc's recursive selection one with explore flag set to True. From a potential point of view, exploring allows to cross forbidden unlikely energy barriers and going out of local minimas.
The following video is an example of refining the position of a molecule using RecursiveGroupSelector and setting refine flag to True. The molecule is always refined from its original position towards a new one generated by the move generator.
The following video is an example of exploring the space of a molecule using RecursiveGroupSelector and setting explore flag to True. The molecule explores the allowed space by wandering via its move generator and only moves enhancing the structure are stored.
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Core.GroupSelector import RecursiveGroupSelector
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# Add constraints ...
# Re-define groups if needed ...
# Re-define groups selector if needed ...
##### Wrap engine group selector with a recursive group selector. #####
# create recursive group selector. Recurrence is set to 20 with explore flag set to True.
RGS = RecursiveGroupSelector(ENGINE.groupSelector, recur=20, refine=False, explore=True)
ENGINE.set_group_selector(RGS)
selector¶The wrapped selector instance.
lastSelectedIndex¶The last selected group index.
willSelect¶Get whether next step a new selection is occur or still the same group is going to be selected again.
willRecur¶Get whether next step the same group will be returned.
willRefine¶Get whether next step the same group will be returned and refine flag is True.
willExplore¶Get whether next step the same group will be returned and explore flag is True.
isNewSelection¶Get whether this last step a new selection was made.
isRecurring¶Get whether this last step the same group was returned.
isRefining¶Get whether this last step the same group was returned and refine flag is True.
isExploring¶Get whether this last step the same group was returned and explore flag is True.
override¶Override flag value.
refine¶Refine flag value.
explore¶Explore flag value.
currentRecur¶The current recur value which is selected group dependant when override flag is True.
recur¶The current recur value. The set recur value can change during engine runtime if override flag is True. To get the recur value as set by set_recur method recurAsSet must be used.
recurAsSet¶Get recur value as set but set_recur method.
position¶Get the position of the selector in the path.
engine¶Get the wrapped selector engine instance.
set_engine(engine)¶Sets the wrapped selector stochastic engine instance.
| Parameters: |
|
|---|
set_recur(recur)¶Sets the recur value.
| Parameters: |
|
|---|
set_override(override)¶Select override value.
| Parameters: |
|
|---|
set_refine(refine)¶Select override value.
| Parameters: |
|
|---|
set_explore(explore)¶Select override value.
| Parameters: |
|
|---|
select_index()¶Select new index.
| Returns: |
|
|---|
MoveGenerator contains parent classes for all move generators. A MoveGenerator sub-class is used at fullrmc’s stochastic engine runtime to generate moves upon selected groups. Every group has its own MoveGenerator class and definitions, therefore it is possible to fully customize how a group of atoms should move.
fullrmc.Core.MoveGenerator.MoveGenerator(group=None)¶Bases: object
It is the parent class of all moves generators. This class can’t be instantiated but its sub-classes might be.
| Parameters: |
|
|---|
group¶Group instance.
set_group(group)¶Set the MoveGenerator group.
| Parameters: |
|
|---|
check_group(group)¶Check the generator’s group. This method must be overloaded in all MoveGenerator sub-classes.
| Parameters: |
|
|---|
transform_coordinates(coordinates, argument=None)¶Transform coordinates. This method is called to move atoms. This method must be overloaded in all MoveGenerator sub-classes.
| Parameters: |
|
|---|---|
| Returns: |
|
move(coordinates)¶Moves coordinates. This method must NOT be overloaded in MoveGenerator sub-classes.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.MoveGenerator.RemoveGenerator(group=None, maximumCollected=None, allowFittingScaleFactor=False, atomsList=None)¶Bases: fullrmc.Core.MoveGenerator.MoveGenerator
This is a very particular move generator that will not generate moves on atoms but removes them from the atomic configuration using a general collector mechanism. Remove generators must be used to create defects in the simulated system. When the standard error is high, removing atoms might reduce the total fit standard error but this can be illusional and very limiting because artificial non physical voids can get created in the system which will lead to an impossibility to finding a solution at the end. It’s strongly recommended to exhaust all ideas and possibilities in finding a good solution prior to start removing atoms unless structural defects is the goal of the simulation.
All removed or amputated atoms are collected by the engine and will become available to be re-inserted in the system if needed. But keep in mind, it might be physically easy to remove and atom but an impossibility to add it back especially if the created voids are smeared out.
Removers are called generators but they behave like selectors. Instead of applying a certain move on a group of atoms, they normally pick atoms from defined atoms list and apply no moves on those. ‘move’ and ‘transform_coordinates’ methods are not implemented in this class of generators and a usage error will be raised if called. ‘pick_from_list’ method is used instead and must be overloaded by all RemoveGenerator subclasses.
N.B. This class can’t be instantiated but its sub-classes might be.
| Parameters: |
|
|---|
atomsList¶Atoms list from which atoms will be picked to attempt removal.
allowFittingScaleFactor¶Whether to allow constraints to fit their scale factor upon removing atoms.
maximumCollected¶Maximum collected atoms allowed.
check_group(group)¶Check the generator’s group.
| Parameters: |
|
|---|
set_maximum_collected(maximumCollected)¶Set maximum collected number of atoms allowed.
| Parameters: |
|
|---|
set_allow_fitting_scale_factor(allowFittingScaleFactor)¶Set allow fitting scale factor flag.
| Parameters: |
|
|---|
set_atoms_list(atomsList)¶Set atoms index list from which atoms will be picked to attempt removal. This method must be overloaded and not be called from this class but from its children. Otherwise a usage error will be raised.
| Parameters: |
|
|---|
move(coordinates)¶Moves coordinates. This method must NOT be overloaded in MoveGenerator sub-classes.
| Parameters: |
|
|---|
transform_coordinates(coordinates, argument)¶This method must NOT be overloaded in MoveGenerator sub-classes.
| Parameters: |
|
|---|
pick_from_list(engine)¶This method must be overloaded in all RemoveGenerator sub-classes.
| Parameters: |
|
|---|
fullrmc.Core.MoveGenerator.SwapGenerator(group=None, swapLength=1, swapList=None)¶Bases: fullrmc.Core.MoveGenerator.MoveGenerator
It is a particular move generator that instead of generating a move upon a group of atoms, it will exchange the group atom positions with other atoms from a defined swapList. Because the swapList can be big, swapGenerator can be assigned to multiple groups at the same time under the condition of all groups having the same length.
During stochastic engine runtime, whenever a swap generator is encountered, all sophisticated selection recurrence modes such as (refining, exploring) will be reduced to simple recurrence.
This class can’t be instantiated but its sub-classes might be.
| Parameters: |
|
|---|
swapLength¶Swap length.
swapList¶Swap list.
groupAtomsIndexes¶Last selected group atoms index.
swapAtomsIndexes¶Last swap atoms index.
set_swap_length(swapLength)¶Set swap length. it will empty and reset swaplist automatically.
| Parameters: |
|
|---|
set_group(group)¶Set the MoveGenerator group.
| Parameters: |
|
|---|
set_swap_list(swapList)¶Set the swap-list to exchange atoms position from.
| Parameters: |
|
|---|
append_to_swap_list(subList)¶Append a sub list to swap list.
| Parameters: |
|
|---|
get_ready_for_move(engine, groupAtomsIndexes)¶Set the swap generator ready to perform a move. Unlike a normal move generator, swap generators will affect not only the selected atoms but other atoms as well. Therefore at stochastic engine runtime, selected atoms will be extended to all affected atoms by the swap.
This method is called automatically upon stochastic engine runtime to ensure that all affect atoms with the swap are updated.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.MoveGenerator.PathGenerator(group=None, path=None, randomize=False)¶Bases: fullrmc.Core.MoveGenerator.MoveGenerator
PathGenerator is a MoveGenerator sub-class where moves definitions are pre-stored in a path and get pulled out at every move step.
This class can’t be instantiated but its sub-classes might be.
| Parameters: |
|
|---|
step¶Current step number.
path¶Path list of moves.
randomize¶Randomize flag.
check_path(path)¶Check the generator’s path.
This method must be overloaded in all PathGenerator sub-classes.
| Parameters: |
|
|---|
normalize_path(path)¶Normalizes all path moves. It is called automatically upon set_path method is called.
This method can be overloaded in all MoveGenerator sub-classes.
| Parameters: |
|
|---|---|
| Returns: |
|
set_path(path)¶Set the moves path.
| Parameters: |
|
|---|
set_randomize(randomize)¶Set whether to randomize moves selection.
| Parameters: |
|
|---|
move(coordinates)¶Move coordinates.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.MoveGenerator.MoveGeneratorCombinator(group=None, combination=None, shuffle=False)¶Bases: fullrmc.Core.MoveGenerator.MoveGenerator
MoveGeneratorCombinator combines all moves of a list of MoveGenerators and applies it at once.
| Parameters: |
|
|---|
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Core.MoveGenerator import MoveGeneratorCombinator
from fullrmc.Generators.Translations import TranslationGenerator
from fullrmc.Generators.Rotations import RotationGenerator
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# Add constraints ...
# Re-define groups if needed ...
##### Define each group move generator as a combination of a translation and a rotation. #####
# create recursive group selector. Recurrence is set to 20 with explore flag set to True.
# shuffle is set to True which means that at every selection the order of move generation
# is random. At one step a translation is performed prior to rotation and in another step
# the rotation is performed at first.
# selected from the collector.
for g in ENGINE.groups:
# create translation generator
TMG = TranslationGenerator(amplitude=0.2)
# create rotation generator only when group length is bigger than 1.
if len(g)>1:
RMG = RotationGenerator(amplitude=2)
MG = MoveGeneratorCombinator(collection=[TMG,RMG],shuffle=True)
else:
MG = MoveGeneratorCombinator(collection=[TMG],shuffle=True)
g.set_move_generator( MG )
shuffle¶Shuffle flag.
combination¶Combination list of MoveGenerator instances.
check_group(group)¶Checks the generator’s group. This methods always returns True because normally all combination MoveGenerator instances groups are checked.
This method must NOT be overloaded unless needed.
| Parameters: |
|
|---|
set_group(group)¶Set the MoveGenerator group.
| Parameters: |
|
|---|
set_combination(combination)¶Set the generators combination list.
| Parameters: |
|
|---|
set_shuffle(shuffle)¶Set whether to shuffle moves generator.
| Parameters: |
|
|---|
move(coordinates)¶Move coordinates.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.MoveGenerator.MoveGeneratorCollector(group=None, collection=None, randomize=True, weights=None)¶Bases: fullrmc.Core.MoveGenerator.MoveGenerator
MoveGeneratorCollector collects MoveGenerators instances and applies the move of one instance at every step.
| Parameters: |
|
|---|
# import fullrmc modules
from fullrmc.Engine import Engine
from fullrmc.Core.MoveGenerator import MoveGeneratorCollector
from fullrmc.Generators.Translations import TranslationGenerator
from fullrmc.Generators.Rotations import RotationGenerator
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb('system.pdb')
# Add constraints ...
# Re-define groups if needed ...
##### Define each group move generator as a combination of a translation and a rotation. #####
# create recursive group selector. Recurrence is set to 20 with explore flag set to True.
# randomize is set to True which means that at every selection a generator is randomly
# selected from the collector.
for g in ENGINE.groups:
# create translation generator
TMG = TranslationGenerator(amplitude=0.2)
# create rotation generator only when group length is bigger than 1.
if len(g)>1:
RMG = RotationGenerator(amplitude=2)
MG = MoveGeneratorCollector(collection=[TMG,RMG],randomize=True)
else:
MG = MoveGeneratorCollector(collection=[TMG],randomize=True)
g.set_move_generator( MG )
randomize¶Randomize flag.
collection¶List of MoveGenerator instances.
generatorsWeight¶Generators selection weights list.
selectionScheme¶Selection scheme.
set_group(group)¶Set the MoveGenerator group.
| Parameters: |
|
|---|
check_group(group)¶Check the generator’s group. This methods always returns True because normally all collection MoveGenerator instances groups are checked.
This method must NOT be overloaded unless needed.
| Parameters: |
|
|---|
set_collection(collection)¶Set the generators instances collection list.
| Parameters: |
|
|---|
set_randomize(randomize)¶Set whether to randomize MoveGenerator instance selection from collection list.
| Parameters: |
|
|---|
set_weights(weights)¶Set groups selection weighting scheme.
| Parameters: |
|
|---|
set_selection_scheme()¶Set selection scheme.
move(coordinates)¶Move coordinates.
| Parameters: |
|
|---|---|
| Returns: |
|
fullrmc.Core.MoveGenerator.generate_random_float()¶random() -> x in the interval [0, 1).
This is a C compiled module to compute boundary conditions related calculations
fullrmc.Core.boundary_conditions_collection.get_reciprocal_basis()¶Computes reciprocal box matrix.
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.boundary_conditions_collection.transform_coordinates()¶Transforms coordinates array using a transformation matrix.
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.boundary_conditions_collection.box_coordinates_real_distances()¶Computes atomic real distances given box coordinates.
| Arguments: |
|
|---|---|
| Returns: |
|
This is a C compiled module to compute transformations from real to reciprocal space and vice versa.
fullrmc.Core.reciprocal_space.gr_to_sq()¶Transform pair correlation function g(r) to static structure factor S(q).
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.reciprocal_space.Gr_to_sq()¶Transform pair distribution function G(r) to static structure factor S(q).
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.reciprocal_space.sq_to_Gr()¶Transform static structure factor S(q) to pair distribution function G(r).
| Arguments: |
|
|---|---|
| Returns: |
|
This is a C compiled module to compute atomic pair distances.
fullrmc.Core.pairs_distances.from_to_points_differences()¶Compute point to point vector difference between two atomic coordinates arrays taking into account periodic or infinite boundary conditions. Difference is calculated as the following:
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_distances.pairs_differences_to_point()¶Compute differences between one atomic coordinates arrays to a point coordinates taking into account periodic or infinite boundary conditions. Difference is calculated as the following:
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_distances.pairs_differences_to_indexcoords()¶Compute differences between one atomic coordinates arrays to a point coordinates given its index in the coordinates array and taking into account periodic or infinite boundary conditions. Difference is calculated as the following:
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_distances.pairs_differences_to_multi_points()¶Compute differences between one atomic coordinates arrays to a multiple points coordinates taking into account periodic or infinite boundary conditions. Difference is calculated as the following:
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_distances.pairs_differences_to_multi_indexcoords()¶Compute differences between one atomic coordinates arrays to a points coordinates given their indexes in the coordinates array and taking into account periodic or infinite boundary conditions. Difference is calculated as the following:
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_distances.pairs_distances_to_point()¶Compute distances between one atomic coordinates arrays to a point coordinates taking into account periodic or infinite boundary conditions. Distances is calculated as the following:
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_distances.pairs_distances_to_indexcoords()¶Compute distances between one atomic coordinates arrays to a points coordinates given their indexes in the coordinates array and taking into account periodic or infinite boundary conditions. Distances is calculated as the following:
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_distances.pairs_distances_to_multi_points()¶Compute distances between one atomic coordinates arrays to a multiple points coordinates taking into account periodic or infinite boundary conditions. Distances is calculated as the following:
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_distances.pairs_distances_to_multi_indexcoords()¶Compute distances between one atomic coordinates arrays to a points coordinates given their indexes in the coordinates array and taking into account periodic or infinite boundary conditions. Distances is calculated as the following:
| Arguments: |
|
|---|---|
| Returns: |
|
This is a C compiled module to compute atomic bonds.
fullrmc.Core.atomic_coordination.single_atom_single_shell_subdists()¶fullrmc.Core.atomic_coordination.single_atom_single_shell_totdists()¶fullrmc.Core.atomic_coordination.single_atom_single_shell_coords()¶fullrmc.Core.atomic_coordination.single_atom_multi_shells_totdists()¶fullrmc.Core.atomic_coordination.single_atom_multi_shells_coords()¶fullrmc.Core.atomic_coordination.single_atom_coord_number_totdists()¶fullrmc.Core.atomic_coordination.single_atom_coord_number_coords()¶fullrmc.Core.atomic_coordination.multi_atoms_coord_number_totdists()¶fullrmc.Core.atomic_coordination.multi_atoms_coord_number_coords()¶fullrmc.Core.atomic_coordination.all_atoms_coord_number_totdists()¶fullrmc.Core.atomic_coordination.all_atoms_coord_number_coords()¶This is a C compiled module to compute atomic inter-molecular distances.
fullrmc.Core.atomic_distances.single_atomic_distances_dists()¶Computes the inter-molecular distances constraint of a single atom given a distances array.
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.atomic_distances.multiple_atomic_distances_coords()¶Computes multiple atoms inter-molecular distances constraint given coordinates.
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.atomic_distances.multiple_atomic_distances_dists()¶Computes multiple atoms inter-molecular distances constraint given distances.
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.atomic_distances.full_atomic_distances_coords()¶Computes all atoms inter-molecular distances constraint given coordinates.
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.atomic_distances.full_atomic_distances_dists()¶Computes all atoms inter-molecular distances constraint given distances.
| Arguments: |
|
|---|---|
| Returns: |
|
This is a C compiled module to compute atomic bonds.
fullrmc.Core.bonds.full_bonds_coords()¶It calculates the bonds constraint of box coordinates.
| Arguments: |
|
|---|---|
| Returns: |
|
This is a C compiled module to compute bonded atoms angle.
fullrmc.Core.angles.full_angles_coords()¶Computes the angles constraint given bonded atoms vectors.
| Arguments: |
|
|---|---|
| Returns: |
|
This is a C compiled module to compute improper angles.
fullrmc.Core.dihedral_angles.full_dihedral_angles_coords()¶Computes the improper angles constraint between an improper atom and a plane atoms. The plane normal vector is calculated using the right-hand rule where (thumb=ox vector), (index=oy vector) hence (oz=normal=second finger)
| Arguments: |
|
|---|---|
| Returns: |
|
This is a C compiled module to compute improper angles.
fullrmc.Core.improper_angles.full_improper_angles_coords()¶Computes the improper angles constraint between an improper atom and a plane atoms. The plane normal vector is calculated using the right-hand rule where (thumb=ox vector), (index=oy vector) hence (oz=normal=second finger)
| Arguments: |
|
|---|---|
| Returns: |
|
This is a C compiled module to compute pair distances histograms.
fullrmc.Core.pairs_histograms.single_pairs_histograms()¶Computes the pair distribution histograms of a single atom given a distances array.
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_histograms.multiple_pairs_histograms_coords()¶Computes the pair distribution histograms of multiple atoms given atomic coordinates.
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_histograms.multiple_pairs_histograms_dists()¶Computes the pair distribution histograms of multiple atoms given atomic distances.
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_histograms.full_pairs_histograms_coords()¶Computes the pair distribution histograms of multiple atoms given atomic coordinates.
| Arguments: |
|
|---|---|
| Returns: |
|
fullrmc.Core.pairs_histograms.full_pairs_histograms_dists()¶Computes the pair distribution histograms of multiple atoms given atomic distances.
| Arguments: |
|
|---|---|
| Returns: |
|