Bachir Aoun; Fullrmc, a Rigid Body Reverse Monte Carlo Modeling Package Enabled with Machine Learning and Artificial Intelligence; J. Comput. Chem. (2016), 37, 1102–1111
Bachir Aoun; Stochastic atomic modeling and optimization with fullrmc; J. Appl. Cryst. (2022). 55, 1664-1676
Molecular system fullrmc stochastic fitting simulation. Groups are set to molecules and smart moves are applied. Translations along symmetry axes, rotations about symmetry axes, etc. |
Atomic binary Nickel-Titanium shape memory alloy system phase transformation stochastic simulation. Random atomic translations are enough to reproduce short range ordering. But swapping atoms is necessary to fit long range atomic correlations. |
Molecular system mere atomic stochastic simulation. Covalent bond electron density polarization is modelled by allowing fullrmc to explore across energy low correlation barriers. |
Reverse Monte Carlo traditional fitting mode compared with fullrmc’s recursive selection with exploring. This video shows how from a potential point of view exploring allow to cross forbidden unlikely barriers and going out of local minimas. |
Reverse Monte Carlo (RMC) is probably best known for its applications in condensed matter physics and solid state chemistry. fullrmc which stands for FUndamental Library Language for Reverse Monte Carlo is different than traditional RMC but a stochastic modelling method to solve an inverse problem whereby an atomic/molecular model is adjusted until its atoms position have the greatest consistency with a set of experimental data.
fullrmc is a python package with its core and calculation modules optimized and compiled in Cython. fullrmc is not a standard RMC package but it’s rather unique in its approach to stochastically solving an atomic or molecular structure. fullrmc’s Engine sub-module is the main module that contains the definition of ‘Engine’ which is the main and only class used to launch a stochastic fitting calculation. Engine reads only Protein Data Bank formatted atomic configuration ‘.pdb’ files and handles other definitions and attributes such as:
- Group: Engine doesn’t understand atoms or molecules but group of atom indexes instead. A group is a set of atom indexes, allowing indexes redundancy within the same group definition. A Group instance can contain any list of indexes and as many atoms index as needed. Grouping atoms is essential to make clusters of atoms (residues, molecules, etc) evolve and move together. A group of a single atom index can be used to make a single atom move separately from the others. Engine’s ‘groups’ attribute is a simple list of group instances containing all the desired and defined groups that one wants to move.
- Group selector: Engine requires a GroupSelector instance which is the artist that selects a group from the engine’s groups list at every engine runtime step. Among other properties, depending on which group selector is used by the engine, a GroupSelector can allow weighting which means selecting groups more or less frequently than the others, it can also allow selection recurrence and refinement of a single group, ordered and random selection is also possible.
- Move generator: Every group instance has its own MoveGenerator. Therefore every group of atoms when selected by the engine’s group selector at the engine’s runtime can perform a customizable and different kind of moves.
- Constraint: A constraint is a rule that controls certain aspect of the configuration upon moving groups. Engine’s ‘constraints’ attribute is a list of all defined and used constraint instances, it is the judge that controls the evolution of the system by accepting or rejecting the move of a group. If engine’s constraints list is empty and contains no constraint definition, this will result in accepting all the generated moves.
## Tetrahydrofuran (THF) molecule sketch
##
## O
## H41 / \ H11
## \ / \ /
## H42-- C4 THF C1 --H12
## \ MOLECULE /
## \ /
## H31-- C3-------C2 --H21
## / \
## H32 H22
##
# ##################################################################################### #
# ############################### IMPORT WHAT IS NEEDED ############################### #
import os
import numpy as np
from fullrmc.Engine import Engine
from fullrmc.Constraints.PairDistributionConstraints import PairDistributionConstraint
from fullrmc.Constraints.DistanceConstraints import InterMolecularDistanceConstraint
from fullrmc.Constraints.BondConstraints import BondConstraint
from fullrmc.Constraints.AngleConstraints import BondsAngleConstraint
from fullrmc.Constraints.ImproperAngleConstraints import ImproperAngleConstraint
from fullrmc.Core.MoveGenerator import MoveGeneratorCollector
from fullrmc.Generators.Translations import TranslationGenerator, TranslationAlongSymmetryAxisGenerator
from fullrmc.Generators.Rotations import RotationGenerator, RotationAboutSymmetryAxisGenerator
# ##################################################################################### #
# ############################# DECLARE USEFUL VARIABLES ############################## #
pdfData = "thf_pdf.exp"
pdbStructure = "thf.pdb"
enginePath = "thf_engine.rmc"
# ##################################################################################### #
# ############################## CREATE STOCHASTIC ENGINE ############################# #
ENGINE = Engine(path=None)
# if engine is not created and saved
if not ENGINE.is_engine(enginePath)
# create and initialize engine
ENGINE = Engine(path=enginePath, freshStart=True)
ENGINE.set_pdb(pdbFileName)
# re-set structure boundary conditions
ENGINE.set_boundary_conditions(np.array([48.860,0,0, 0,48.860,0, 0,0,48.860]))
# create and add pair distribution constraint to the engine
PDF_CONSTRAINT = PairDistributionConstraint(experimentalData=pdfData, weighting="atomicNumber")
ENGINE.add_constraints([PDF_CONSTRAINT])
# create and add intermolecular distances constraint to the engine
EMD_CONSTRAINT = InterMolecularDistanceConstraint()
ENGINE.add_constraints([EMD_CONSTRAINT])
# create and add bonds constraint to the engine
B_CONSTRAINT = BondConstraint()
ENGINE.add_constraints([B_CONSTRAINT])
B_CONSTRAINT.create_bonds_by_definition( bondsDefinition={"THF": [('O' ,'C1' , 1.20, 1.70),
('O' ,'C4' , 1.20, 1.70),
('C1','C2' , 1.25, 1.90),
('C2','C3' , 1.25, 1.90),
('C3','C4' , 1.25, 1.90),
('C1','H11', 0.88, 1.16),('C1','H12', 0.88, 1.16),
('C2','H21', 0.88, 1.16),('C2','H22', 0.88, 1.16),
('C3','H31', 0.88, 1.16),('C3','H32', 0.88, 1.16),
('C4','H41', 0.88, 1.16),('C4','H42', 0.88, 1.16)] })
# create and add angles constraint to the engine
BA_CONSTRAINT = BondsAngleConstraint()
ENGINE.add_constraints([BA_CONSTRAINT])
BA_CONSTRAINT.create_angles_by_definition( anglesDefinition={"THF": [ ('O' ,'C1' ,'C4' , 105, 125),
('C1' ,'O' ,'C2' , 100, 120),
('C4' ,'O' ,'C3' , 100, 120),
('C2' ,'C1' ,'C3' , 95 , 115),
('C3' ,'C2' ,'C4' , 95 , 115),
# H-C-H angle
('C1' ,'H11','H12', 98 , 118),
('C2' ,'H21','H22', 98 , 118),
('C3' ,'H31','H32', 98 , 118),
('C4' ,'H41','H42', 98 , 118),
# H-C-O angle
('C1' ,'H11','O' , 100, 120),
('C1' ,'H12','O' , 100, 120),
('C4' ,'H41','O' , 100, 120),
('C4' ,'H42','O' , 100, 120),
# H-C-C
('C1' ,'H11','C2' , 103, 123),
('C1' ,'H12','C2' , 103, 123),
('C2' ,'H21','C1' , 103, 123),
('C2' ,'H21','C3' , 103, 123),
('C2' ,'H22','C1' , 103, 123),
('C2' ,'H22','C3' , 103, 123),
('C3' ,'H31','C2' , 103, 123),
('C3' ,'H31','C4' , 103, 123),
('C3' ,'H32','C2' , 103, 123),
('C3' ,'H32','C4' , 103, 123),
('C4' ,'H41','C3' , 103, 123),
('C4' ,'H42','C3' , 103, 123) ] })
# create and add improper angles constraint to the engine keeping THF molecules' atoms in the plane
IA_CONSTRAINT = ImproperAngleConstraint()
ENGINE.add_constraints([IA_CONSTRAINT])
IA_CONSTRAINT.create_angles_by_definition( anglesDefinition={"THF": [ ('C2','O','C1','C4', -15, 15),
('C3','O','C1','C4', -15, 15) ] })
# initialize constraints data
ENGINE.initialize_used_constraints()
# save engine
ENGINE.save()
# if engine is created and saved, it can be simply loaded.
else:
ENGINE = ENGINE.load(engineFilePath)
# unpack constraints before fitting in case tweaking is needed
PDF_CONSTRAINT, EMD_CONSTRAINT, B_CONSTRAINT, BA_CONSTRAINT, IA_CONSTRAINT = ENGINE.constraints
# ##################################################################################### #
# ########################### RUN ATOMIC STOCHASTIC FITTING ########################### #
# set groups as atoms. By default when the engine is constructed, all groups are single atoms.
ENGINE.set_groups_as_atoms()
ENGINE.run(numberOfSteps=100000, saveFrequency=1000)
# ##################################################################################### #
# ########################## RUN MOLECULAR STOCHASTIC FITTING ######################### #
## set groups as molecules instead of atoms
ENGINE.set_groups_as_molecules()
# set moves generators to all groups as a collection of random translation and rotation
for g in ENGINE.groups:
mg = MoveGeneratorCollector(collection=[TranslationGenerator(),RotationGenerator()], randomize=True)
g.set_move_generator( mg )
## Uncomment to use any of the following moves generators instead of the earlier collector
## Also other moves generators can be used to achieve a better fit for instance:
#[g.set_move_generator(TranslationAlongSymmetryAxisGenerator(axis=0)) for g in ENGINE.groups]
#[g.set_move_generator(TranslationAlongSymmetryAxisGenerator(axis=1)) for g in ENGINE.groups]
#[g.set_move_generator(TranslationAlongSymmetryAxisGenerator(axis=2)) for g in ENGINE.groups]
#[g.set_move_generator(RotationAboutSymmetryAxisGenerator(axis=0)) for g in ENGINE.groups]
#[g.set_move_generator(RotationAboutSymmetryAxisGenerator(axis=1)) for g in ENGINE.groups]
#[g.set_move_generator(RotationAboutSymmetryAxisGenerator(axis=2)) for g in ENGINE.groups]
## Molecular constraints are not necessary any more because groups are set to molecules.
## At every engine step a whole molecule is rotate or translated therefore its internal
## distances and properties are safe from any changes. At any time constraints can be
## turn on again using the same method with a True flag. e.g. B_CONSTRAINT.set_used(True)
B_CONSTRAINT.set_used(False)
BA_CONSTRAINT.set_used(False)
IA_CONSTRAINT.set_used(False)
## run engine and perform stochastic fitting on molecules
ENGINE.run(numberOfSteps=100000, saveFrequency=1000)
# ##################################################################################### #
# ################################## PLOT CONSTRAINTS ################################# #
PDF_CONSTRAINT.plot(show=False)
EMD_CONSTRAINT.plot(show=False)
B_CONSTRAINT.plot(lineWidth=2, nbins=20, split='element', show=False)
BA_CONSTRAINT.plot(lineWidth=2, nbins=20, split='element', show=False)
IA_CONSTRAINT.plot(lineWidth=2, nbins=20, split='element', show=True )
The result shown in the figures herein is obtained by running fullrmc Engine for several hours on molecular groups. Position optimization is achieved by using a RecursiveGroupSelector to refine every selected group position and alternating groups move generators. RotationAboutSymmetryAxisGenerator is used to fit the ring orientation, then TranslationAlongSymmetryAxisGenerator is used to translate molecules along meaningful directions. At the end, reset groups to single atom index and RandomSelector is used to select groups randomly. the Engine is run for additional several hours to refine atoms positions separately.
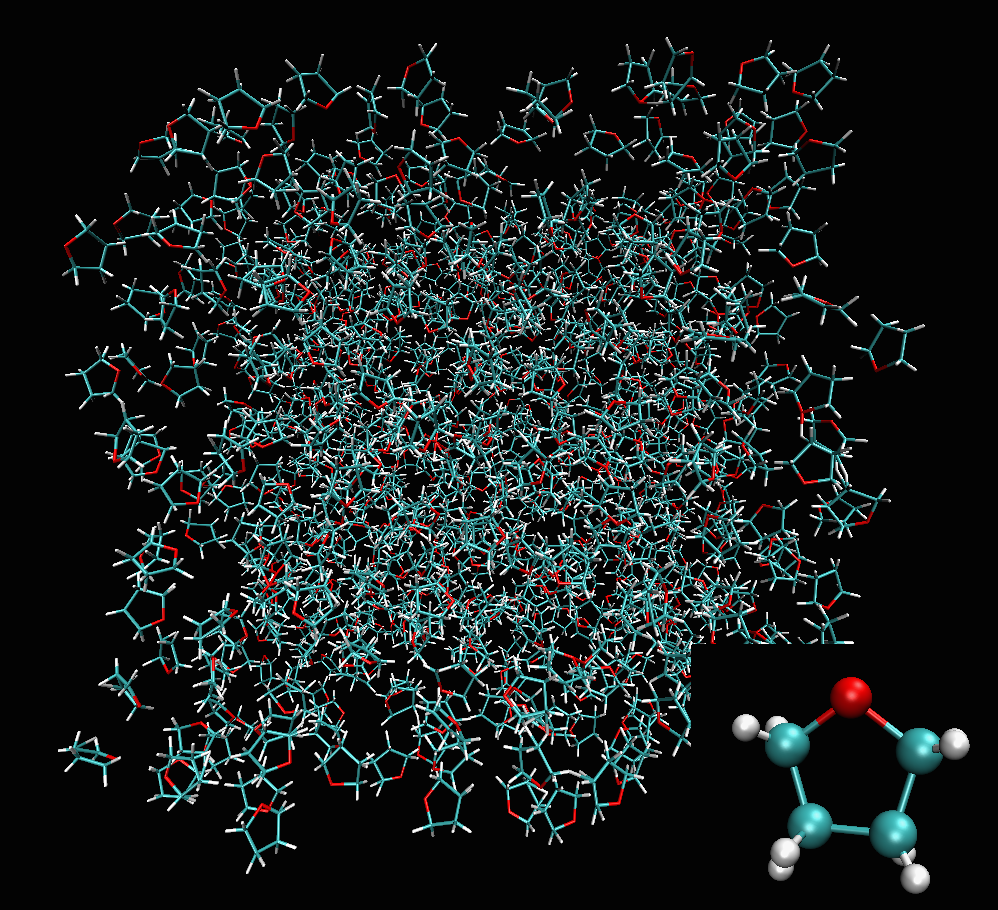
a) Structure containing 800 Tetrahydrofuran randomly generated. |
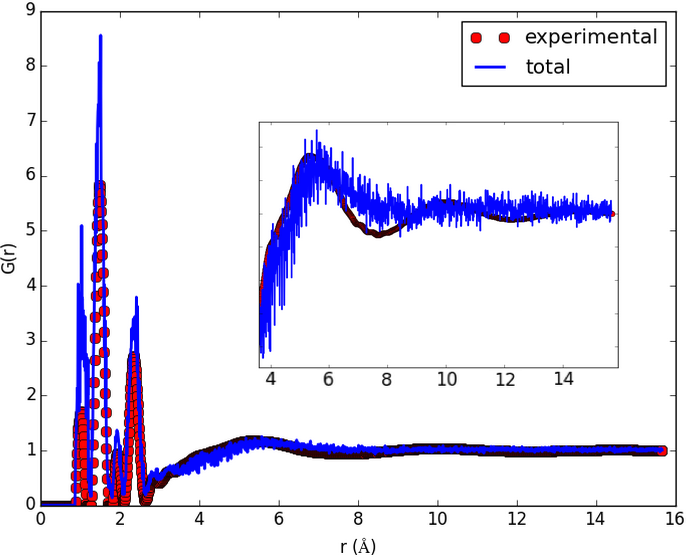
b) Initial pair distribution function calculated before any fitting. |
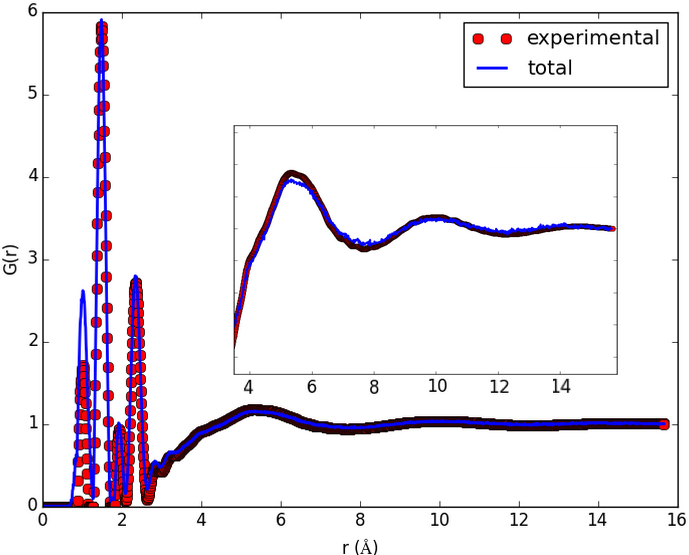
c) pair distribution function calculated after about 20 hours of Engine runtime. |
Engine is fullrmc’s main module. It contains ‘Engine’ the main class of fullrmc which is the stochastic artist. The engine class takes only Protein Data Bank formatted files ‘.pdb’ as atomic/molecular input structure. It handles and fits simultaneously many experimental data while controlling the evolution of the system using user-defined molecular and atomistic constraints such as bond-length, bond-angles, dihedral angles, inter-molecular-distances, etc.
fullrmc.Engine.InterceptHook(path, password=None)Bases: object
Engine runtime intercept hook. This can be used by a thread or another process to intercept a running stochastic engine to do different actions such as stop, save, export a pdb file or reset the standard error.
Calling InterceptHook methods on a non-running engine won’t do any action.
| Parameters: |
|
|---|
From a different python process, run the following code to handle a hook of a running engine.
# import fullrmc's InterceptHook
from fullrmc import InterceptHook
# create hook
hook = InterceptHook(path='my_engine.rmc')
# safely force stopping running engine
hook.stop_engine()
# safely force saving running engine
hook.save_engine()
# safely export a pdb file of the current engine structure status
hook.export_pdb()
# safely reset standard error during engine runtime
hook.reset_standard_error()
stop_engine()stop a running engine
save_engine()force saving a running engine
export_pdb()export a pdb of the current state of a running engine
reset_standard_error()reset a running engine total standard error
get_engine_hook(engine)Get engine instance intercept hook
clear(engine)release all unexecuted hooks from engine
fullrmc.Engine.Engine(path=None, logFile=None, freshStart=False, timeout=10, password=None)Bases: object
fulrmc’s engine, is used to launch a stochastic modelling which is different than traditional Reverse Monte Carlo (RMC). It has the capability to use and fit simultaneously multiple sets of experimental data. One can also define constraints such as distances, bonds length, angles and many others.
| Parameters: |
|
|---|
# import engine
from fullrmc.Engine import Engine
# create engine
ENGINE = Engine(path='my_engine.rmc')
# set pdb file
ENGINE.set_pdb(pdbFileName)
# Add constraints ...
# Re-define groups if needed ...
# Re-define groups selector if needed ...
# Re-define moves generators if needed ...
# save engine
ENGINE.save()
# run engine for 10000 steps and save only at the end
ENGINE.run(numberOfSteps=10000, saveFrequency=10000)
codify(export='codified_engine.py', name='ENGINE', engineSaveName='engine.rmc', frames=None, addDependencies=True)Codify engine full state to a code that once executed it will regenerate and and save the engine on disk. This is a better alternative to transfering engine from one system to another. The generated code is python 2 and 3 compatible and operating system safe allowing to get an executable code to regenerate the engine.
| Parameters: |
|
|---|---|
| Returns: |
|
pathEngine’s repository path if set or save.
versionStochastic engine version.
infoStochastic engine information (version, id) tuple.
framesStochastic engine frames list copy.
framesPathList of engine traditional frames name and multiframes path.
usedFrameStochatic engine frame in use.
lastSelectedGroupIndexThe last moved group instance index in groups list.
lastSelectedGroupThe last moved group instance.
lastSelectedAtomsIndexThe last moved atoms index.
stateEngine’s state.
generatedNumber of generated moves.
removedRemoved atoms tuple (tried, accepted, ratio)
triedNumber of tried moves.
acceptedNumber of accepted moves.
toleratedNumber of tolerated steps in spite of increasing totalStandardError
toleranceTolerance in percent.
groupsEngine’s defined groups list.
pdbEngine’s pdbparser instance.
boundaryConditionsEngine’s boundaryConditions instance.
isPBCWhether boundaryConditions are periodic.
isIBCWhether boundaryConditions are infinte.
basisVectorsThe boundary conditions basis vectors in case of PeriodicBoundaries, None in case of InfiniteBoundaries.
reciprocalBasisVectorsThe boundary conditions reciprocal basis vectors in case of PeriodicBoundaries, None in case of InfiniteBoundaries.
volumeThe boundary conditions basis volume in case of PeriodicBoundaries, None in case of InfiniteBoundaries.
realCoordinatesThe real coordinates of the current configuration.
boxCoordinatesThe box coordinates of the current configuration in case of PeriodicBoundaries. Similar to realCoordinates in case of InfiniteBoundaries.
numberOfMoleculesNumber of molecules.
moleculesIndexAtoms molecule index list.
moleculesNameAtoms molecule name list.
elementsIndexAtoms element index list indexing elements sorted set.
elementsSorted set of all existing atom elements.
allElementsAtoms element list.
namesIndexAtoms name index list indexing names sorted set
namesSorted set of all existing atom names.
allNamesAtoms name list.
numberOfNamesLength of atoms name set.
numberOfAtomsNumber of atoms in the pdb.
numberOfAtomsPerNameNumber of atoms per name dictionary.
numberOfElementsNumber of different elements in the pdb.
numberOfAtomsPerElementNumber of atoms per element dictionary.
numberDensitySystem’s number density computed as \(\rho_{0}=\frac{N}{V}\) where N is the total number of atoms and V the volume of the system.
constraintsCopy list of all constraints instances.
groupSelectorEngine’s group selector instance.
totalStandardErrorEngine’s last recorded totalStandardError of the current configuration.
timeout()Timeout to successfully acquire the lock upon reading or writing to the repository
set_timeout(timeout)Set repository access timeout
| Parameters: |
|
|---|
get_original_data(name, frame=None)Get original data as initialized and parsed from pdb.
| Parameters: |
|
|---|---|
| Returns: |
|
is_engine(path, repo=False, mes=False, safeMode=True)Get whether a fullrmc engine is stored in the given path.
| Parameters: |
|
|---|---|
| Returns: |
|
save(path=None, copyFrames=True)Save engine to disk.
| Parameters: |
|
|---|
N.B. If path is given, it will automatically update engine’s path to point towards given path.
load(path, safeMode=True)Load and return engine instance. None of the current engine attribute will be updated. must be used as the following:
# import engine
from fullrmc.Engine import Engine
# create engine
ENGINE = Engine().load(path)
| Parameters: |
|
|---|---|
| Returns: |
|
set_log_file(logFile)Set the log file basename.
| Parameters: |
|
|---|
is_frame(frame)Check whether a given frame exists.
| Parameters: |
|
|---|---|
| Returns: |
|
add_frames(frames, create=False)Add a one or many (multi)frame to engine.
| Parameters: |
|
|---|
add_frame(*args, **kwargs)alias to add_frames
reinit_frame(frame)Reset frame data to initial pdb coordinates.
| Parameters: |
|
|---|
get_frame_category(frame)Get whether a given frame name is a normal frame or a multiframe or a multiframe subframe. If frame does not exist an error will be raised.
| Parameters: |
|
|---|---|
| Returns: |
|
set_used_frame(frame, _updateRepo=True)Switch engine frame.
| Parameters: |
|
|---|
use_frame(*args, **kwargs)alias to set_used_frame.
delete_frame(frame)Delete frame data from Engine as well as from repository .
| Parameters: |
|
|---|
rename_frame(frame, newName)Rename (multi)frame.
| Parameters: |
|
|---|
export_pdb(path, frame=None)Export a pdb file of the last refined and saved configuration state.
| Parameters: |
|
|---|
get_pdb(frame=None)Get a pdb instance of the last refined and save configuration state.
| Parameters: |
|
|---|---|
| Returns: |
|
set_tolerance(tolerance)Set engine’s runtime tolerance value.
| Parameters: |
|
|---|
set_group_selector(selector, frame=None)Set engine’s group selector instance.
| Parameters: |
|
|---|
clear_groups()Clear all engine’s defined groups.
remove_groups(groups)Remove groups by instance, name or index
| Parameters: |
|
|---|
add_group(g, name=None, _check=True)Add a group to engine’s groups list.
| Parameters: |
|
|---|
set_groups(groups, name='group', add=False, _check=True)Set engine’s groups.
| Parameters: |
|
|---|
add_groups(groups, name=None, _check=True)Add groups to engine.
| Parameters: |
|
|---|
THIS METHOD WILL BE DEPRECATED. Must use ‘set_groups’ instead with ‘add’ flag set to True
set_groups_as_atoms(name='atom', add=False)Automatically creates groups as single atom group for all atoms. If add is True, created groups will be added to engine existing ones otherwise old groups will be removed.
| Parameters: |
|
|---|
set_groups_as_molecules(name='molecule', add=False)Automatically create molecular groups of atom indexes according to molecules indexes. If add is True, created groups will be added to engine existing ones otherwise old groups will be removed.
| Parameters: |
|
|---|
set_pdb(pdb, boundaryConditions=None, names=None, elements=None, moleculesIndex=None, moleculesName=None)Set used frame pdb configuration. Engine and constraints data will be automatically reset but not constraints definitions. If pdb was already set and this is a resetting to a different atomic configuration, with different elements or atomic order, or different size and number of atoms, constraints definitions must be reset manually. In general, their is no point in changing the atomic configuration of a completely different atomic nature. It is advisable to create a new engine from scratch or redefining all constraints definitions.
| Parameters: |
|
|---|
set_boundary_conditions(boundaryConditions, _broadcast=True)Sets the configuration’s boundary conditions. Any type of periodic or infinite boundary conditions is allowed and not restricted to cubic. Engine and constraints data will be automatically reset. Number density will be automatically calculated upon setting boundary conditions. In the case where inifinite boundaries are set, which is needed to simulate isolated atomic systems such as nano-particles, the volume is theoretically infinite and therefore number density must be 0. But experimentally the measured samples are diluted in solvants and the actual number density must be the experimental one. To avoid numerical instabilities, number density will be automatically set to water’s one equal to 0.0333679 and volume will be adjusted to the ratio of number of atoms devided to the given water number density. If this number is not accurate, user can always set the appropriate number density using the stochastic engine ‘set_number_density’ method
| Parameters: |
|
|---|
set_number_density(numberDensity)Sets system’s number density. This is used to correct system’s volume. It can only be used with InfiniteBoundaries.
| Parameters: |
|
|---|
set_molecules_index(moleculesIndex=None, moleculesName=None, _broadcast=True)Set moleculesIndex list, assigning each atom to a molecule.
| Parameters: |
|
|---|
set_elements_index(elements=None, _broadcast=True)Set elements and elementsIndex lists, assigning a type element to each atom.
| Parameters: |
|
|---|
set_names_index(names=None, _broadcast=True)Set names and namesIndex list, assigning a name to each atom.
| Parameters: |
|
|---|
visualize(frame=None, commands=None, foldIntoBox=False, boxToCenter=False, boxWidth=2, boxStyle='solid', boxColor='yellow', bgColor='black', displayParams=None, representationParams='Lines', otherParams=None)Visualize the last configuration using pdbparser visualize_vmd method.
| Parameters: |
|
|---|
add_constraints(constraints, allSubframes=False)Add constraints to the engine. If used frame is a normal frame, all other normal frames will have the constraints added. If used frame is a subframe, all other subframes of the same multiframe will have the experimental constraints added to. But given non-experimental constraints will be only added to the used frame.
| Parameters: |
|
|---|
remove_constraints(constraints, allSubframes=False)Remove constraints from engine list of constraints.
| Parameters: |
|
|---|
reset_constraints()Reset used normal frame constraints flags.
reset_engine()Re-initialize engine and resets constraints flags and data.
compute_total_standard_error(constraints, current='standardError')Compute the total standard error as the sum of all constraints’ standard error.
Where:
\(variance_{i}\) is the variance value of the constraint i.
\(stdErr_{i}\) the standard error of the constraint i defined as \(\sum \limits_{j}^{points} (target_{i,j}-computed_{i,j})^{2} = (Y_{i,j}-F(X_{i,j}))^{2}\)
| Parameters: |
|
|---|---|
| Returns: |
|
update_total_standard_error()Compute and set engine’s total totalStandardError of used constraints.
get_used_constraints(sortConstraints=False)Parses all engine’s constraints and returns different lists of the active (used) ones.
| Parameters: |
|
|---|---|
| Returns: |
|
initialize_used_constraints(force=False, sortConstraints=False)Calls get_used_constraints method, re-initializes constraints when needed and return them all.
| Parameters: |
|
|---|---|
| Returns: |
|
run(numberOfSteps=100000, sortConstraints=True, saveFrequency=1000, frame=None, xyzFrequency=None, xyzPath='trajectory.xyz', restartPdb='restart.pdb', ncores=None)Run stochastic fitting engine.
| Parameters: |
|
|---|
plot_constraints(frame=None, constraintsID=None, plotKwargs=None)Plot constraints
fullrmc.Engine.generate_random_float()random() -> x in the interval [0, 1).
This module provides all the global types, variables and some general methods that fullrmc needs.
fullrmc.Globals.range(*args)fullrmc.Globals.Logger(*args, **kwargs)Bases: pysimplelog.SimpleLog.SingleLogger
custom_init()Custom initialize abstract method. This method will be called at the end of initialzation. This method needs to be overloaded to custom initialize Logger instances.
| Parameters: |
|
|---|
fixed(message)alias to message at fixed level
accepted(message)alias to message at move accepted level
rejected(message)alias to message at move rejected level
nottried(message)alias to message at move not tried level
saved(message)alias to message at save engine level
impl(message)alias to message at implement engine level
implement(message)alias to message at usage engine level
usage(message)alias to message at usage engine level
frame(message)alias to message at setting engine level
report(message)alias to message at report engine level